Porfiria
| Porfiria | |
|---|---|
 | |
| La figura de la izquierda es la orina del primer día, mientras que la figura de la derecha es la orina después de tres días de exposición al sol y muestra el clásico cambio de color a púrpura. | |
| Pronunciación | |
| Especialidad | Hematología , dermatología , neurología. |
| Síntomas | Dependiendo del subtipo: dolor abdominal , dolor en el pecho , vómitos , confusión, estreñimiento , fiebre , convulsiones , ampollas con la luz solar [1] [2] |
| Inicio habitual | Ataques recurrentes que duran días o semanas [2] |
| Causas | Generalmente genético [2] |
| Método de diagnóstico | Análisis de sangre, orina y heces, pruebas genéticas [2] |
| Diagnóstico diferencial | Envenenamiento por plomo , enfermedad hepática alcohólica [3] |
| Tratamiento | Depende del tipo y los síntomas [2] |
| Frecuencia | 1 a 100 en 50.000 personas [1] |
La porfiria / p ɔːr ˈ f ɪr i ə / es un grupo de trastornos en los que las sustancias llamadas porfirinas se acumulan en el cuerpo, afectando negativamente a la piel o al sistema nervioso . [1] Los tipos que afectan al sistema nervioso también se conocen como porfiria aguda, ya que los síntomas aparecen rápidamente y duran poco. [1] Los síntomas de un ataque incluyen dolor abdominal , dolor en el pecho , vómitos , confusión, estreñimiento , fiebre , presión arterial alta y frecuencia cardíaca alta . [1] [2] [4] Los ataques suelen durar días o semanas. [2] Las complicaciones pueden incluir parálisis , niveles bajos de sodio en sangre y convulsiones . [4] Los ataques pueden ser desencadenados por el alcohol , el tabaquismo , los cambios hormonales, el ayuno, el estrés o ciertos medicamentos. [2] [4] Si la piel se ve afectada, pueden aparecer ampollas o picazón con la exposición a la luz solar. [2]
La mayoría de los tipos de porfiria se heredan de uno o ambos padres de una persona y se deben a una mutación en uno de los genes que producen hemo . [2] Pueden heredarse de manera autosómica dominante , autosómica recesiva o dominante ligada al cromosoma X. [1] Un tipo, la porfiria cutánea tardía , también puede deberse a la hemocromatosis (aumento de hierro en el hígado), la hepatitis C , el alcohol o el VIH/SIDA . [1] El mecanismo subyacente resulta en una disminución en la cantidad de hemo producido y una acumulación de sustancias involucradas en la producción de hemo. [1] Las porfirias también pueden clasificarse según si el hígado o la médula ósea están afectados. [1] El diagnóstico generalmente se realiza mediante análisis de sangre, orina y heces. [2] Se pueden realizar pruebas genéticas para determinar la mutación específica. [2] Las porfirias hepáticas son aquellas en las que la deficiencia enzimática ocurre en el hígado. Las porfirias hepáticas incluyen la porfiria intermitente aguda (AIP), la porfiria variegata (VP), la porfiria por deficiencia de deshidratasa del ácido aminolevulínico (ALAD), la coproporfiria hereditaria (HCP) y la porfiria cutánea tarda. [5]
El tratamiento depende del tipo de porfiria y de los síntomas de la persona. [2] El tratamiento de la porfiria de la piel generalmente implica evitar la luz solar, mientras que el tratamiento de la porfiria aguda puede implicar administrar hemo intravenoso o una solución de glucosa . [2] En raras ocasiones, puede realizarse un trasplante de hígado . [2]
La prevalencia precisa de la porfiria no está clara, pero se estima que afecta entre 1 y 100 por cada 50.000 personas. [1] Las tasas son diferentes en todo el mundo. [2] Se cree que la porfiria cutánea tardía es el tipo más común. [1] La enfermedad fue descrita ya en el año 370 a. C. por Hipócrates . [6] El mecanismo subyacente fue descrito por primera vez por el fisiólogo y químico alemán Felix Hoppe-Seyler en 1871. [6] El nombre porfiria proviene del griego πορφύρα, porphyra , que significa " púrpura ", una referencia al color de la orina que puede estar presente durante un ataque. [6]
Signos y síntomas
Esta sección necesita citas adicionales para su verificación . ( Enero de 2016 ) |


Porfirias agudas
Porfiria intermitente aguda (AIP), porfiria variegata (VP), porfiria por deficiencia de deshidratasa del ácido aminolevulínico (ALAD) y coproporfiria hereditaria (HCP). Estas enfermedades afectan principalmente al sistema nervioso , lo que da lugar a crisis episódicas conocidas como ataques agudos. El síntoma principal de un ataque agudo es el dolor abdominal , a menudo acompañado de vómitos , hipertensión (presión arterial elevada) y taquicardia (frecuencia cardíaca anormalmente rápida). [4]
Los episodios más graves pueden implicar complicaciones neurológicas: típicamente neuropatía motora (disfunción grave de los nervios periféricos que inervan los músculos), que conduce a debilidad muscular y potencialmente a cuadriplejia (parálisis de las cuatro extremidades) y síntomas del sistema nervioso central, como convulsiones y coma . Ocasionalmente, puede haber síntomas psiquiátricos de corta duración, como ansiedad, confusión, alucinaciones y, muy raramente, psicosis manifiesta. Todos estos síntomas se resuelven una vez que pasa el ataque agudo. [ cita requerida ]
Dadas las múltiples presentaciones y la incidencia relativamente baja de porfiria, inicialmente se puede sospechar que los pacientes tienen otras afecciones no relacionadas. Por ejemplo, la polineuropatía de la porfiria aguda puede confundirse con el síndrome de Guillain-Barré , y se recomiendan comúnmente las pruebas de porfiria en esas situaciones. [7] La elevación del ácido aminolevulínico debido a la interrupción de la síntesis del hemo inducida por el plomo da como resultado una intoxicación por plomo con síntomas similares a la porfiria aguda. [8] [9] [10] [11] [12] [13]
Porfirias crónicas
Las porfirias no agudas son la protoporfiria dominante ligada al cromosoma X (XLDPP), la porfiria eritropoyética congénita (CEP), la porfiria cutánea tardía (PCT) y la protoporfiria eritropoyética (EPP). Ninguna de ellas se asocia a ataques agudos; su manifestación primaria es la enfermedad cutánea. Por este motivo, estas cuatro porfirias (junto con dos porfirias agudas, VP y HCP, que también pueden implicar manifestaciones cutáneas) se denominan a veces porfirias cutáneas.
Las enfermedades de la piel se producen cuando se acumula un exceso de porfirinas en la piel. Las porfirinas son moléculas fotoactivas y la exposición a la luz hace que los electrones alcancen niveles de energía más altos. Cuando estos vuelven al nivel de energía en reposo o estado fundamental, se libera energía. Esto explica la propiedad de fluorescencia típica de las porfirinas, que causa daño local en la piel.
En la porfiria se observan dos patrones distintos de enfermedad de la piel:
- Fotosensibilidad inmediata. Es típica de la XLDPP y la EPP. Tras un período variable de exposición al sol (normalmente unos 30 minutos), los pacientes se quejan de dolor intenso, ardor y malestar en las zonas expuestas. Normalmente, los efectos no son visibles, aunque ocasionalmente puede haber algo de enrojecimiento e hinchazón de la piel.
- Enfermedad cutánea vesiculoerosiva. Esta (una referencia a las ampollas (vesículas) y llagas abiertas (erosiones) características que se observan en los pacientes) es el patrón observado en CEP, PCT, VP y HCP. Los cambios se notan solo en las áreas expuestas al sol, como la cara y el dorso de las manos. La enfermedad cutánea más leve, como la que se observa en VP y HCP, consiste en una mayor fragilidad de la piel en las áreas expuestas con una tendencia a formar ampollas y erosiones, en particular después de golpes o rasguños menores. Estas se curan lentamente, a menudo dejando pequeñas cicatrices que pueden ser más claras o más oscuras que la piel normal. A veces se observa una enfermedad cutánea más grave en la PCT, con lesiones prominentes, oscurecimiento de la piel expuesta como la cara e hipertricosis : crecimiento anormal del vello en la cara, en particular en las mejillas. La enfermedad más grave se observa en CEP y una variante rara de PCT conocida como porfiria hepatoeritropoyética (HEP); Los síntomas incluyen un acortamiento grave de los dedos, pérdida de apéndices cutáneos como el pelo y las uñas, y cicatrices graves en la piel con desaparición progresiva de las orejas, los labios y la nariz. Los pacientes también pueden presentar dientes deformados y descoloridos o anomalías en las encías y los ojos.
Porfirias congénitas
- Las porfirias congénitas son trastornos genéticos causados por mutaciones en enzimas que participan en la vía de biosíntesis del hemo. Existen varios tipos de porfirias congénitas, entre ellas la protoporfiria eritropoyética (PPE), la porfiria eritropoyética congénita (PEC) y la porfiria cutánea tardía (PCT). Cada tipo se caracteriza por deficiencias enzimáticas específicas que conducen a la acumulación de diferentes porfirinas.
- La protoporfiria eritropoyética (PPE) es causada por una deficiencia de ferroquelatasa, que conduce a la acumulación de protoporfirina IX en los glóbulos rojos, el plasma y los tejidos. Los pacientes con PPE experimentan fotosensibilidad grave y la exposición a la luz solar provoca reacciones cutáneas dolorosas.
- La porfiria eritropoyética congénita (PEC), también conocida como enfermedad de Günther, es consecuencia de una deficiencia de la uroporfirinógeno III sintasa. Esto provoca la acumulación de uroporfirina I y coproporfirina I en la médula ósea, la sangre y la orina. Los síntomas de la PEC incluyen fotosensibilidad grave, anemia, esplenomegalia y, a menudo, lesiones cutáneas desfigurantes.
- El diagnóstico de las porfirias congénitas implica una evaluación clínica, pruebas bioquímicas y análisis genético. El tratamiento tiene como objetivo controlar los síntomas y prevenir los ataques agudos evitando los desencadenantes, como la exposición a la luz solar, ciertos medicamentos y el alcohol. Además, los tratamientos pueden incluir flebotomía para reducir los niveles de hierro en la PCT, administración de preparaciones de hemo para aliviar los síntomas y trasplante de hígado en casos graves. El diagnóstico temprano y el tratamiento adecuado son cruciales para mejorar la calidad de vida de las personas con porfirias congénitas.
Causa
Las porfirias generalmente se consideran de naturaleza genética. [ cita requerida ]
Genética
Los subtipos de porfirias dependen de qué enzima sea deficiente.
| Tipo de porfiria | Enzima deficiente | Tipo de porfiria | Herencia | Síntomas | Predominio |
|---|---|---|---|---|---|
| Porfiria por deficiencia de aminolevulinato deshidratasa (ALADP) | 5-aminolevulinato deshidratasa (ALAD) | Hepático | Autosómica recesiva [14] | Dolor abdominal, neuropatía [14] | Extremadamente raro; menos de 10 casos reportados. [15] |
| Porfiria intermitente aguda (PAI) | Hidroximetilbilano sintasa (HMBS), anteriormente porfobilinógeno desaminasa (PBGD) | Hepático | Autosómico dominante [14] | Dolor abdominal periódico, neuropatía periférica , trastornos psiquiátricos, taquicardia [14] | 1 en 10.000 [16] –20.000 [16] |
| Porfiria eritropoyética congénita (PEC) | uroporfirinógeno sintasa (UROS) | Eritropoyético | Autosómica recesiva [14] | Fotosensibilidad grave con eritema, hinchazón y formación de ampollas. Anemia hemolítica, esplenomegalia [14] | 1 en 1.000.000 o menos. [17] |
| Porfiria cutánea tardía (PCT) | uroporfirinógeno descarboxilasa (UROD) | Hepático | Aproximadamente el 80% esporádico, [18] 20% autosómico dominante [14] | Fotosensibilidad con vesículas y ampollas [14] | 1 en 10.000 [19] |
| Coproporfiria hereditaria (HCP) | coproporfirinógeno oxidasa (CPOX) | Hepático | Autosómico dominante [14] | Fotosensibilidad, síntomas neurológicos, cólicos [14] | 1 en 500.000 [19] |
| Porfiria hardero | coproporfirinógeno oxidasa (CPOX) | Eritropoyético | Autosómica recesiva [14] | Ictericia, anemia, agrandamiento del hígado y del bazo, a menudo neonatal. Fotosensibilidad posterior. | Extremadamente raro; menos de 10 casos reportados. |
| Porfiria variegata (VP) | protoporfirinógeno oxidasa (PPOX) | Hepático | Autosómico dominante [20] | Fotosensibilidad, síntomas neurológicos, retraso del desarrollo. | 1 de cada 300 en Sudáfrica [19] 1 de cada 75.000 en Finlandia [21] |
| Protoporfiria eritropoyética (PPE) | ferroquelatasa (FECH) | Eritropoyético | Autosómica recesiva [14] | Fotosensibilidad con lesiones cutáneas. Cálculos biliares, disfunción hepática leve [14] | 1 en 75.000 [19] –200.000 [19] |
La protoporfiria dominante ligada al cromosoma X es una forma rara de protoporfiria eritropoyética causada por una mutación de ganancia de función en ALAS2 caracterizada por fotosensibilidad grave . [22] [23]
En los tipos autosómicos recesivos, si una persona hereda un solo gen, puede convertirse en portadora. Generalmente no presenta síntomas, pero puede transmitir el gen a su descendencia. [24]
Desencadenantes
La porfiria aguda puede ser desencadenada por varios fármacos, la mayoría de los cuales se cree que la desencadenan al interactuar con enzimas del hígado que se producen con el hemo. Entre estos fármacos se incluyen: [25] [26] [27]
- Sulfonamidas , incluidas sulfadiazina , sulfasalazina y trimetoprima/sulfametoxazol .
- Sulfonilureas como glibenclamida , gliclazida y glimepirida , aunque se cree que la glipizida es segura.
- Barbitúricos, incluidos tiopental , fenobarbital , primidona , etc.
- Tratamiento sistémico con antimicóticos como fluconazol , griseofulvina , ketoconazol y voriconazol . (Se cree que el uso tópico de estos agentes es seguro debido a su mínima absorción sistémica).
- Ciertos antibióticos como rifapentina , rifampicina , rifabutina , isoniazida , nitrofurantoína y, posiblemente, metronidazol .
- Derivados del cornezuelo, entre ellos dihidroergotamina , ergometrina , ergotamina , metisergida , etc.
- Ciertos medicamentos antirretrovirales ( por ejemplo , indinavir , nevirapina , ritonavir , saquinavir , etc.)
- Progestágenos
- Algunos anticonvulsivos incluyen: carbamazepina , etosuximida , fenitoína , topiramato , valproato .
- Algunos analgésicos como el dextropropoxifeno , el ketorolaco , el metamizol y la pentazocina.
- Algunos tratamientos contra el cáncer como bexaroteno , busulfán , clorambucilo , estramustina , etopósido , flutamida , idarrubicina , ifosfamida , irinotecán , ixabepilona , letrozol , lomustina , megestrol , mitomicina , mitoxantrona , paclitaxel , procarbazina , tamoxifeno , topotecán
- Algunos antidepresivos como la imipramina , la fenelzina y la trazodona.
- Algunos antipsicóticos como la risperidona y la ziprasidona.
- Algunos retinoides utilizados para afecciones de la piel como la acitretina y la isotretinoína.
- Otros varios, incluidos: cocaína , metildopa , fenfluramina , disulfiram , orfenadrina , pentoxifilina y aurotiomalato de sodio .
Patogenesia
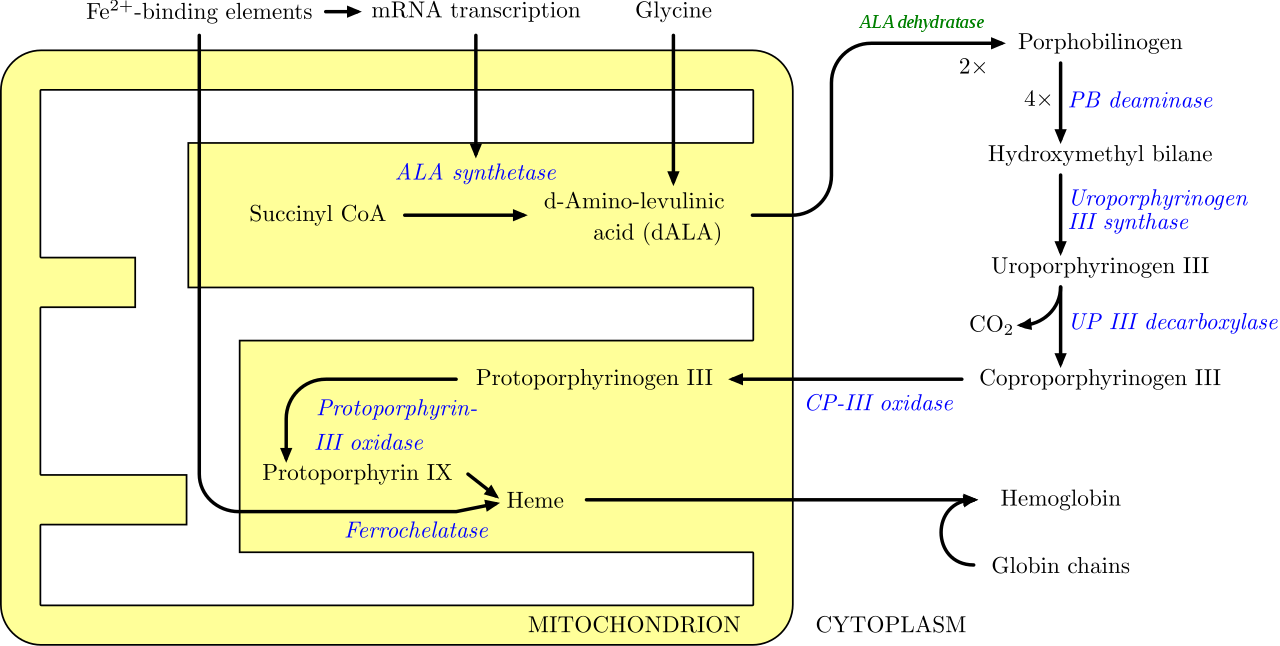
En los seres humanos , las porfirinas son los principales precursores del hemo , un componente esencial de la hemoglobina , la mioglobina , la catalasa , la peroxidasa y los citocromos hepáticos P450 . [28]
El cuerpo requiere de porfirinas para producir hemo , que se utiliza para transportar oxígeno en la sangre entre otras cosas, pero en las porfirias hay una deficiencia (hereditaria o adquirida) de las enzimas que transforman las diversas porfirinas en otras, lo que lleva a niveles anormalmente altos de una o más de estas sustancias. Las porfirias se clasifican de dos formas, por los síntomas y por la fisiopatología. Fisiológicamente, las porfirias se clasifican en hepáticas o eritropoyéticas en función de los sitios de acumulación de los precursores del hemo , ya sea en el hígado o en la médula ósea y los glóbulos rojos . [29]
La deficiencia de las enzimas de la vía de las porfirinas conduce a una producción insuficiente de hemo . La función del hemo desempeña un papel central en el metabolismo celular . Este no es el principal problema en las porfirias; la mayoría de las enzimas de síntesis del hemo , incluso las enzimas disfuncionales , tienen suficiente actividad residual para ayudar en la biosíntesis del hemo . El principal problema en estas deficiencias es la acumulación de porfirinas , los precursores del hemo , que son tóxicos para los tejidos en altas concentraciones. Las propiedades químicas de estos intermediarios determinan la ubicación de la acumulación, si inducen fotosensibilidad y si el intermediario se excreta (en la orina o las heces ). [ cita requerida ]
Hay ocho enzimas en la vía biosintética del hemo , cuatro de las cuales (la primera y las tres últimas) se encuentran en las mitocondrias , mientras que las otras cuatro se encuentran en el citosol . Los defectos en cualquiera de ellas pueden provocar alguna forma de porfiria. Las porfirias hepáticas se caracterizan por ataques neurológicos agudos ( convulsiones , psicosis , dolor abdominal y de espalda extremo y polineuropatía aguda ), mientras que las formas eritropoyéticas se presentan con problemas cutáneos, generalmente una erupción ampollosa sensible a la luz y un aumento del crecimiento del cabello . La porfiria variegata (también porfiria variegata o porfiria mixta ), que resulta de una deficiencia parcial de la PROTO oxidasa , se manifiesta con lesiones cutáneas similares a las de la porfiria cutánea tarda combinadas con ataques neurológicos agudos. La coproporfiria hereditaria , que se caracteriza por una deficiencia de la coproporfirinógeno oxidasa, codificada por el gen CPOX, también puede presentarse con ataques neurológicos agudos y lesiones cutáneas. Todas las demás porfirias predominan en la piel o en los nervios. [30]
Diagnóstico
Estudios de porfirina
La porfiria se diagnostica mediante análisis bioquímicos de sangre , orina y heces . [17] [31] En general, la estimación de porfobilinógeno (PBG) en orina es el primer paso si se sospecha porfiria aguda. Como resultado de la retroalimentación, la disminución de la producción de hemo conduce a un aumento de la producción de precursores, siendo PBG una de las primeras sustancias en la vía de síntesis de porfirina. [32] En casi todos los casos de síndromes de porfiria aguda, el PBG urinario está notablemente elevado excepto en la muy rara deficiencia de ALA deshidratasa o en pacientes con síntomas debido a tirosinemia hereditaria tipo I. [ 33] En casos de porfiria inducida por envenenamiento por mercurio o arsénico , aparecen otros cambios en los perfiles de porfirina, más notablemente elevaciones de uroporfirinas I y III, coproporfirinas I y III y precoproporfirina. [34]
Como la mayoría de las porfirias son enfermedades poco frecuentes , los laboratorios de los hospitales generales no suelen tener la experiencia, la tecnología ni el tiempo del personal para realizar pruebas de porfiria. En general, las pruebas implican enviar muestras de sangre, heces y orina a un laboratorio de referencia. [17] Todas las muestras para detectar porfirinas deben manipularse adecuadamente. Las muestras deben tomarse durante un ataque agudo; de lo contrario, puede producirse un resultado falso negativo . Las muestras deben protegerse de la luz y refrigerarse o conservarse. [17]
Si todos los estudios de porfirinas son negativos, se debe considerar la posibilidad de una pseudoporfiria . Una revisión cuidadosa de la medicación a menudo permitirá encontrar la causa de la pseudoporfiria. [ cita requerida ]
Pruebas adicionales
Es posible que se requieran otras pruebas diagnósticas de los órganos afectados, como estudios de conducción nerviosa para neuropatía o una ecografía del hígado. Las pruebas bioquímicas básicas pueden ayudar a identificar enfermedades hepáticas , carcinoma hepatocelular y otros problemas orgánicos. [35]
•Otros diagnósticos
Evaluación clínica: una historia clínica completa y un examen físico centrado en los síntomas relacionados con la fotosensibilidad, lesiones cutáneas, dolor abdominal y manifestaciones neurológicas.
Pruebas genéticas: Pruebas genéticas moleculares para identificar mutaciones genéticas específicas asociadas con porfirias congénitas.
Otras pruebas: Se pueden realizar pruebas de función hepática, estudios de hierro y estudios de imágenes como ecografía o resonancia magnética para evaluar la afectación del hígado y el bazo.
Gestión
Porfiria aguda
Administración de carbohidratos
A menudo, se requiere tratamiento empírico si la sospecha diagnóstica de una porfiria es alta, ya que los ataques agudos pueden ser fatales. Por lo general, se recomienda una dieta rica en carbohidratos; en ataques graves, se inicia una infusión de dextrosa al 10%, que puede ayudar en la recuperación al suprimir la síntesis de hemo , lo que a su vez reduce la tasa de acumulación de porfirina. Sin embargo, esto puede empeorar los casos de niveles bajos de sodio en sangre ( hiponatremia ) y debe realizarse con extrema precaución, ya que puede resultar fatal. [36]
Análogos del hemo
La hematina (nombre comercial Panhematin) y el hemo arginato (nombre comercial NormoSang) son los fármacos de elección en la porfiria aguda, en los Estados Unidos y el Reino Unido, respectivamente. Estos fármacos deben administrarse muy temprano en un ataque para que sean efectivos; la efectividad varía entre individuos. No son fármacos curativos pero pueden acortar los ataques y reducir la intensidad de un ataque. Los efectos secundarios son raros pero pueden ser graves. Estas sustancias similares al hemo inhiben teóricamente la ALA sintasa y, por lo tanto, la acumulación de precursores tóxicos. En el Reino Unido, los suministros de NormoSang se mantienen en dos centros nacionales; el St Thomas's Hospital , Londres, ofrece suministros de emergencia. [37] En los Estados Unidos, Lundbeck fabrica y suministra Panhematin para infusión. [38]
El hemo arginato (NormoSang) se utiliza durante las crisis, pero también como tratamiento preventivo para evitarlas, un tratamiento cada 10 días. [ cita requerida ]
Cualquier signo de hiponatremia o debilidad debe tratarse con la adición de hematina, hemo arginato o incluso mesoporfirina de estaño , ya que son signos de un síndrome inminente de secreción inadecuada de hormona antidiurética (SIADH) o de un compromiso del sistema nervioso periférico que puede ser localizado o grave y progresar a paresia bulbar y parálisis respiratoria. [ cita requerida ]
Cimetidina
También se ha informado que la cimetidina es eficaz para la crisis porfírica aguda y posiblemente eficaz para la profilaxis a largo plazo. [39]
Control de síntomas
El dolor es intenso, con frecuencia desproporcionado con respecto a los signos físicos, y a menudo requiere el uso de opiáceos para reducirlo a niveles tolerables. El dolor debe tratarse lo antes posible desde el punto de vista médico. Las náuseas pueden ser intensas; pueden responder a los fármacos fenotiazínicos , pero a veces son intratables. Los baños y duchas calientes pueden reducir las náuseas temporalmente, aunque se debe tener precaución para evitar quemaduras o caídas. [ cita requerida ]
Identificación temprana
Se recomienda que los pacientes con antecedentes de porfiria aguda, e incluso los portadores genéticos, lleven una pulsera de alerta u otra identificación en todo momento. Esto es así en caso de que presenten síntomas graves o en caso de accidentes en los que exista un potencial de exposición a medicamentos y, como resultado, no puedan explicar su condición a los profesionales de la salud. Algunos medicamentos están absolutamente contraindicados para pacientes con cualquier forma de porfiria. [40]
Trastornos neurológicos y psiquiátricos
Los pacientes que experimentan ataques frecuentes pueden desarrollar dolor neuropático crónico en las extremidades, así como dolor crónico en el abdomen . [41] La pseudoobstrucción intestinal , el íleo , la intususcepción , la hipoganglionosis y la encopresis en niños se han asociado con porfirias. Se cree que esto se debe al deterioro del nervio axonal en las áreas afectadas del sistema nervioso y a la disfunción del nervio vago. El tratamiento del dolor con opioides de acción prolongada , como la morfina , suele estar indicado y, en los casos en que hay convulsiones o neuropatía, se sabe que la gabapentina mejora el resultado. [42]
Las convulsiones suelen acompañar a esta enfermedad. La mayoría de los medicamentos anticonvulsivos exacerban esta afección. El tratamiento puede ser problemático: se deben evitar especialmente los barbitúricos . Algunas benzodiazepinas son seguras y, cuando se utilizan junto con medicamentos anticonvulsivos más nuevos, como la gabapentina, ofrecen un posible régimen para el control de las convulsiones. La gabapentina tiene la característica adicional de ayudar en el tratamiento de algunos tipos de dolor neuropático. [42] El sulfato de magnesio y los bromuros también se han utilizado en las convulsiones porfirias; sin embargo, el desarrollo de estado epiléptico en la porfiria puede no responder al magnesio solo. La adición de hematina o arginato de hemo se ha utilizado durante el estado epiléptico . [43]
La depresión suele acompañar a la enfermedad y la mejor forma de tratarla es tratar los síntomas que la provocan y, si es necesario, utilizar de forma prudente antidepresivos . Algunos fármacos psicotrópicos son porfirinógenos, lo que limita el alcance terapéutico. Pueden aparecer otros síntomas psiquiátricos como ansiedad, inquietud, insomnio, depresión, manía, alucinaciones, delirios, confusión, catatonía y psicosis. [44]
Enfermedad hepática subyacente
Algunas enfermedades hepáticas pueden causar porfiria incluso en ausencia de predisposición genética. Entre ellas se encuentran la hemocromatosis y la hepatitis C. Puede ser necesario el tratamiento de la sobrecarga de hierro. [2]
Los pacientes con porfirias agudas ( AIP , HCP , VP ) tienen un mayor riesgo a lo largo de su vida de padecer carcinoma hepatocelular (cáncer hepático primario) y pueden requerir seguimiento. No es necesario que estén presentes otros factores de riesgo típicos del cáncer hepático. [2]
Tratamiento hormonal
Las fluctuaciones hormonales que contribuyen a los ataques cíclicos en las mujeres se han tratado con anticonceptivos orales y hormonas luteinizantes para interrumpir los ciclos menstruales. Sin embargo, los anticonceptivos orales también han desencadenado fotosensibilidad y la retirada de los anticonceptivos orales ha desencadenado ataques. Los andrógenos y las hormonas de la fertilidad también han desencadenado ataques. [45] En 2019, el givosiran fue aprobado en los Estados Unidos para el tratamiento de la porfiria hepática aguda. [46] [47]
Porfiria eritropoyética
Estas se asocian con la acumulación de porfirinas en los eritrocitos y son raras.
El dolor, el ardor, la hinchazón y la picazón que se producen en las porfirias eritropoyéticas (EP) generalmente requieren evitar la luz solar intensa. La mayoría de los tipos de protector solar no son efectivos, pero las camisas de manga larga con factor de protección solar, los sombreros, los pañuelos y los guantes pueden ayudar. Se puede utilizar cloroquina para aumentar la secreción de porfirina en algunas EP. [17] En ocasiones, se utiliza la transfusión de sangre para suprimir la producción innata de hemo. [ cita requerida ]
La más rara es la porfiria eritropoyética congénita (PEC), también conocida como enfermedad de Gunther . Los signos pueden presentarse desde el nacimiento e incluyen fotosensibilidad grave, dientes marrones que fluorescen con luz ultravioleta debido a la deposición de porfirinas tipo 1 y, más tarde, hipertricosis . Generalmente se desarrolla anemia hemolítica. Se puede utilizar betacaroteno de calidad farmacéutica en su tratamiento. [48] Un trasplante de médula ósea también ha tenido éxito en la curación de la PEC en algunos casos, aunque aún no se dispone de resultados a largo plazo. [49]
En diciembre de 2014, la afamelanotida recibió la autorización de la Comisión Europea como tratamiento para la prevención de la fototoxicidad en pacientes adultos con PPE. [50] En un ensayo de fase 2 financiado por la industria en 2023, se informó que el dersimelagon, un agonista selectivo del receptor de melanocortina 1 administrado por vía oral que aumenta los niveles de eumelanina en la piel, aumentó la duración de la exposición a la luz solar sin síntomas y la calidad de vida en comparación con placebo en pacientes con protoporfiria eritropoyética. [51]
Epidemiología
Se ha estimado que las tasas de todos los tipos de porfiria en conjunto son aproximadamente una en 25.000 en los Estados Unidos. [52] Se ha estimado que la prevalencia mundial es de entre una en 500 y una en 50.000 personas. [53]
Se han detectado porfirias en todas las razas y en múltiples grupos étnicos de todos los continentes. Hay informes de alta incidencia de AIP en áreas de la India y Escandinavia. Se conocen más de 200 variantes genéticas de AIP, algunas de las cuales son específicas de familias, aunque se ha demostrado que algunas cepas son mutaciones repetidas. [ cita requerida ]
Otra información
La epidemiología de las porfirias congénitas varía según el tipo específico de porfiria. A continuación, se ofrece una descripción general:
1. Protoporfiria eritropoyética (PPE): la PPE es relativamente rara, con una prevalencia estimada de 1 a 9 casos por cada 100.000 personas en todo el mundo. Afecta tanto a hombres como a mujeres y suele presentarse en la infancia o en la adultez temprana.
2. Porfiria eritropoyética congénita (PEC): la PEC es extremadamente rara, con menos de 200 casos reportados en todo el mundo. Se hereda de manera autosómica recesiva, lo que significa que ambos padres deben ser portadores de un gen mutado para que un niño desarrolle la enfermedad. La PEC se presenta con mayor frecuencia en ciertas poblaciones, incluidas las personas de ascendencia del norte de Europa.
3. Porfiria cutánea tardía (PCT): la PCT es la forma más común de porfiria, con una prevalencia estimada de 1 a 2 casos por cada 10.000 personas en la población general. Afecta predominantemente a los adultos, con una prevalencia mayor en hombres que en mujeres. La PCT puede ser esporádica o familiar y suele estar asociada a una enfermedad hepática subyacente, abuso de alcohol, infección por hepatitis C o ciertos medicamentos.
Estas estimaciones de prevalencia pueden variar en distintas regiones y poblaciones, y la prevalencia real de las porfirias congénitas puede estar subnotificada debido a dificultades para el diagnóstico y la concienciación. Además, los avances en las pruebas genéticas y una mayor concienciación sobre la porfiria pueden conducir a datos epidemiológicos más precisos en el futuro.
Historia
El mecanismo subyacente fue descrito por primera vez por el fisiólogo alemán Felix Hoppe-Seyler en 1871, [54] y las porfirias agudas fueron descritas por el médico holandés Barend Stokvis en 1889. [55] [56]
Los vínculos entre las porfirias y las enfermedades mentales se han observado durante décadas. A principios de la década de 1950, los pacientes con porfirias (a veces denominadas "hemofilia porfírica" [57] ) y síntomas graves de depresión o catatonia eran tratados con terapia de electroshock .
Vampiros y hombres lobo
Se ha sugerido la porfiria como una explicación del origen de las leyendas de vampiros y hombres lobo , basándose en ciertas similitudes percibidas entre la condición y el folclore .
En enero de 1964, el artículo de L. Illis de 1963, "Sobre la porfiria y la etiología de los hombres lobo", se publicó en Proceedings of the Royal Society of Medicine . Más tarde, Nancy Garden defendió una conexión entre la porfiria y la creencia en los vampiros en su libro de 1973, Vampires . En 1985, el artículo del bioquímico David Dolphin para la Asociación Estadounidense para el Avance de la Ciencia , "Porfiria, vampiros y hombres lobo: la etiología de las leyendas europeas de la metamorfosis", obtuvo una amplia cobertura mediática, popularizando la idea. [ cita requerida ]
La teoría ha sido rechazada por algunos folcloristas e investigadores por no describir con precisión las características de las leyendas originales de hombres lobo y vampiros o la enfermedad, y por estigmatizar potencialmente a las personas con porfiria. [58] [59]
Un artículo de 1995 del Postgraduate Medical Journal (a través del NIH ) explica:
Como se creía que el vampiro folclórico podía moverse libremente durante las horas del día, a diferencia de la variante del siglo XX, la porfiria eritropoyética congénita no puede explicar fácilmente el vampiro folclórico, pero puede ser una explicación del vampiro tal como lo conocemos en el siglo XX. Además, el vampiro folclórico, cuando se desenterró, siempre se describió como bastante saludable ("como era en vida"), mientras que debido a los aspectos desfigurantes de la enfermedad, los afectados no habrían pasado la prueba de exhumación. Las personas con porfiria eritropoyética congénita no anhelan la sangre. La enzima (hematina) necesaria para aliviar los síntomas no se absorbe intacta en la ingestión oral, y beber sangre no tendría ningún efecto beneficioso en el paciente. Finalmente, y lo más importante, el hecho de que los informes sobre vampiros fueran literalmente rampantes en el siglo XVIII, y que la porfiria eritropoyética congénita sea una manifestación extremadamente rara de una enfermedad rara, la convierte en una explicación poco probable del vampiro folclórico. [60]
Casos notables
- Jorge III . La enfermedad mental que exhibió Jorge III en la crisis de la regencia de 1788 ha inspirado varios intentos de diagnóstico retrospectivo . El primero, escrito en 1855, treinta y cinco años después de su muerte, concluyó que tenía manía aguda . M. Guttmacher, en 1941, sugirió la psicosis maníaco-depresiva como un diagnóstico más probable. La primera sugerencia de que una enfermedad física era la causa del trastorno mental del rey Jorge llegó en 1966, en un artículo llamado "La locura del rey Jorge III: un caso clásico de porfiria", [61] con un seguimiento en 1968, "Porfiria en las casas reales de Estuardo, Hannover y Prusia". [62] Los artículos, por un equipo de psiquiatras madre/hijo , fueron escritos como si el caso de la porfiria hubiera sido probado, pero la respuesta demostró que muchos expertos, incluidos los más íntimamente familiarizados con las manifestaciones de la porfiria, no estaban convencidos. Muchos psiquiatras no estuvieron de acuerdo con el diagnóstico, sugiriendo que el trastorno bipolar era mucho más probable. La teoría se trata en Purple Secret, [63] que documenta la búsqueda finalmente infructuosa de evidencia genética de porfiria en los restos de miembros de la realeza sospechosos de haberla tenido. [64] En 2005, se sugirió que el arsénico (que se sabe que es porfirógeno) administrado a Jorge III con antimonio podría haber causado su porfiria. [65] Este estudio encontró altos niveles de arsénico en el cabello del rey Jorge. En 2010, un análisis de registros históricos argumentó que la afirmación de porfiria se basaba en una interpretación espuria y selectiva de fuentes médicas e históricas contemporáneas. [66] La enfermedad mental de Jorge III es la base de la trama de La locura del rey Jorge , una película británica de 1994 basada en la obra de teatro de Alan Bennett de 1991 , La locura de Jorge III . Los créditos finales de la película incluyen el comentario de que los síntomas del rey sugieren que tenía porfiria, y señalan que la enfermedad es "periódica, impredecible y hereditaria". El argumento tradicional de que Jorge III no tenía porfiria, sino trastorno bipolar, es defendido a fondo por Andrew Roberts en su nueva biografía El último rey de América . [67]
- Descendientes de Jorge III. Entre otros descendientes de Jorge III que los autores de El secreto púrpura teorizaron que padecían porfiria (basándose en el análisis de su extensa y detallada correspondencia médica) se encontraban su tataranieta, la princesa Carlota de Prusia ( la hermana mayor del emperador Guillermo II ) y su hija, la princesa Feodora de Sajonia-Meiningen . Descubrieron mejor evidencia de que el tataranieto de Jorge III, el príncipe Guillermo de Gloucester, fue diagnosticado de manera confiable con porfiria variegata. [68]
- María, reina de Escocia . Se cree que María, reina de Escocia, antepasada del rey Jorge III , también padecía porfiria intermitente aguda, [69] aunque esto es objeto de mucho debate. Se supone que heredó el trastorno, si es que lo padecía, de su padre, Jacobo V de Escocia . Tanto el padre como la hija sufrieron ataques bien documentados que podrían enmarcarse en la constelación de síntomas de porfiria.
- María I de Portugal . También se cree que María I, conocida como «María la Piadosa» o «María la Loca» debido tanto a su fervor religioso como a su aguda enfermedad mental, que la hizo incapaz de manejar los asuntos de estado después de 1792, tuvo porfiria. Francis Willis , el mismo médico que trató a Jorge III, incluso fue convocado por la corte portuguesa, pero regresó a Inglaterra después de que la corte limitara los tratamientos que podía supervisar. Fuentes contemporáneas, como el secretario de Estado de Asuntos Exteriores Luís Pinto de Sousa Coutinho , señalaron que la reina tenía dolores de estómago y espasmos abdominales cada vez más graves: características distintivas de la porfiria. [70]
- Vlad III Drácula, "El Empalador". También se decía que Vlad III sufría de porfiria aguda, lo que pudo haber dado origen a la idea de que los vampiros eran alérgicos a la luz solar. [71]
- Vincent van Gogh . Otros comentaristas han sugerido que Vincent van Gogh pudo haber sufrido porfiria intermitente aguda. [72]
- Rey Nabucodonosor de Babilonia . La descripción de este rey en Daniel 4 sugiere para algunos que padecía porfiria.
- El médico Archie Cochrane nació con porfiria, lo que le causó problemas de salud durante toda su vida. [73]
- Paula Frías Allende . Hija de la novelista chilena Isabel Allende . Cayó en un coma inducido por porfiria en 1991, [74] lo que inspiró a Isabel a escribir las memorias Paula , dedicadas a ella.
- María, reina de Escocia, c. 1578


_-_Google_Cultural_Institute.jpg/1280px-Maria_I,_Queen_of_Portugal_-_Giuseppe_Troni,_atribu%C3%ADdo_(Turim,_1739-Lisboa,_1810)_-_Google_Cultural_Institute.jpg)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
_-_Google_Cultural_Institute.jpg){kind=link}
Usos en la literatura
En algunas publicaciones, en particular en la literatura gótica, se incluyen referencias explícitas o implícitas a la porfiria, como las siguientes:
- La condición es el nombre del personaje principal del poema gótico " El amante de Porfiria ", de Robert Browning .
- Se da a entender que esta enfermedad es la causa de los síntomas que sufre el narrador en el cuento gótico " Lusus Naturae ", de Margaret Atwood . Algunos de los síntomas del narrador se parecen a los de la porfiria, y un pasaje del relato afirma que el nombre de la enfermedad del narrador "tenía algunas P y R". [ cita requerida ]
Referencias
- ^ abcdefghijk "porfiria". MedlinePlus . Julio de 2009 . Consultado el 31 de marzo de 2021 .
- ^ abcdefghijklmnopqr «Porfiria | NIDDK». Instituto Nacional de Diabetes y Enfermedades Digestivas y Renales . Consultado el 6 de diciembre de 2018 .
- ^ Dancygier, Henryk (2009). Hepatología clínica: principios y práctica de las enfermedades hepatobiliares. Springer Science & Business Media. pág. 1088. ISBN 9783642045196Archivado desde el original el 8 de septiembre de 2017.
- ^ abcd Stein, PE; Badminton, MN; Rees, DC (febrero de 2017). "Actualización de la revisión de las porfirias agudas". British Journal of Haematology . 176 (4): 527–538. doi : 10.1111/bjh.14459 . PMID 27982422. S2CID 19970176.
- ^ Kothadia, Jiten P.; LaFreniere, Kilian; Shah, Jamil M. (2024), "Porfiria hepática aguda", StatPearls , Treasure Island (FL): StatPearls Publishing, PMID 30725863 , consultado el 31 de marzo de 2024
- ^ abc McManus, Linda (2014). Patobiología de las enfermedades humanas: una enciclopedia dinámica de los mecanismos de las enfermedades. Elsevier. p. 1488. ISBN 9780123864574Archivado desde el original el 8 de septiembre de 2017.
- ^ Albers JW, Fink JK (2004). "Neuropatía porfírica" (PDF) . Muscle Nerve . 30 (4): 410–422. doi :10.1002/mus.20137. hdl : 2027.42/34640 . PMID: 15372536. S2CID : 68067335.
- ^ Vannotti A (1954). Porfirinas: su importancia biológica y química. Hilger & Watts, Hilger Division. p. 126.
De hecho, el envenenamiento por plomo, como todas las enfermedades porfirínicas, se acompaña de estreñimiento persistente, lesiones nerviosas, hiperpigmentación y ataques abdominales.
- ^ Dancygier H (2009). Hepatología clínica: principios y práctica de las enfermedades hepatobiliares. Springer Science & Business Media. pág. 1088. ISBN 9783642045196Archivado desde el original el 8 de septiembre de 2017.
- ^ Akshatha LN, Rukmini MS, Mamatha TS, Sadashiva Rao P, Prashanth B (diciembre de 2014). "¡Envenenamiento por plomo que imita la porfiria aguda!". Journal of Clinical and Diagnostic Research . 8 (12): CD01-2. doi :10.7860/JCDR/2014/10597.5315. PMC 4316248 . PMID 25653942.
- ^ Tsai MT, Huang SY, Cheng SY (2017). "El envenenamiento por plomo puede ser fácilmente diagnosticado erróneamente como porfiria aguda y dolor abdominal inespecífico". Informes de casos en medicina de emergencia . 2017 : 9050713. doi : 10.1155/2017/9050713 . PMC 5467293. PMID 28630774 .
- ^ Wang, B.; Bissell, DM; Adam, MP; Ardinger, HH; Pagon, RA; Wallace, SE; Bean LJH; Stephens, K.; Amemiya, A. (2018). "Coproporfiria hereditaria". GeneReviews . PMID 23236641. Consultado el 28 de febrero de 2020.
Los síntomas de intoxicación por plomo imitan estrechamente los de la porfiria aguda
. - ^ Murphy, Brian (3 de noviembre de 2020). "¿Qué pruebas pueden ayudar a diagnosticar la porfiria?" . Consultado el 22 de mayo de 2021 .
- ^ abcdefghijklm Tabla 18-1 en: Marks, Dawn B.; Swanson, Todd; Sandra I Kim; Marc Glucksman (2007). Bioquímica y biología molecular . Filadelfia: Wolters Kluwer Health/Lippincott Williams & Wilkins. ISBN 978-0-7817-8624-9.
- ^ Descripción general de las porfirias Archivado el 22 de julio de 2011 en Wayback Machine en The Porphyrias Consortium (una parte de la Red de investigación clínica de enfermedades raras (RDCRN) del NIH) Consultado en junio de 2011
- ^ ab Medscape - Enfermedades del metabolismo del tetrapirrol - Refsum Enfermedad y porfirias hepáticas Archivado el 4 de junio de 2011 en Wayback Machine Autor: Norman C Reynolds. Editor jefe: Stephen A Berman. Actualizado: 23 de marzo de 2009
- ^ abcde Thadani H, Deacon A, Peters T (2000). "Diagnóstico y tratamiento de la porfiria". BMJ . 320 (7250): 1647–1651. doi :10.1136/bmj.320.7250.1647. PMC 1127427 . PMID 10856069.
- ^ "Entrada OMIM - 176090 - PORFIRIA CUTÁNEA TARDA, TIPO I". omim.org . Consultado el 24 de febrero de 2021 .
- ^ abcde Arceci, Robert.; Hann, Ian M.; Smith, Owen P. (2006). Hematólogo pediátrico . Malden, Mass.: Blackwell Pub. ISBN 978-1-4051-3400-2.
- ^ Singal, AK; Anderson, KE; Pagon, RA; Adam, MP; Ardinger, HH; Wallace, SE; Amemiya, A; Bean, LJH; Bird, TD; Dolan, CR; Fong, CT; Smith, RJH; Stephens, K (2013). "Porfiria variegada". GeneReviews . Seattle, EE. UU.: Universidad de Washington. PMID 23409300. Archivado desde el original el 18 de enero de 2017. Consultado el 18 de abril de 2015 .
- ^ Mustajoki P (1980). "Porfiria variegata. Doce años de experiencia en Finlandia". The Quarterly Journal of Medicine . 49 (194): 191–203. PMID 7433635.
- ^ "Entrada OMIM - # 300752 - PROTOPORFIRIA, ERITROPOYÉTICA, LIGADA AL X; XLEPP". omim.org .
- ^ "Orphanet: Protoporfiria eritropoyética ligada al cromosoma X". www.orpha.net .
- ^ "Porfiria - Síntomas y causas". Mayo Clinic . Consultado el 5 de marzo de 2020 .
- ^ Tishler, PV. "Acerca de la porfiria: base de datos de seguridad". Base de datos de seguridad de medicamentos para la porfiria . Fundación Estadounidense de Porfiria. Archivado desde el original el 12 de julio de 2017. Consultado el 27 de agosto de 2017 .
- ^ Brun, A. "napos". Base de datos de fármacos para la porfiria aguda . Red Europea de Porfiria. Archivado desde el original el 23 de agosto de 2017. Consultado el 27 de agosto de 2017 .
- ^ Comité Conjunto de Formularios (2013). Formulario Nacional Británico (BNF) (65.ª ed.). Londres, Reino Unido: Pharmaceutical Press. p. 663. ISBN 978-0-85711-084-8.
- ^ Kumari, Asha (12 de octubre de 2017). Sweet Biochemistry. Elsevier Science. ISBN 9780128144534. Recuperado el 30 de octubre de 2022 .
- ^ Lourenço, Charles Marquez; Lee, Chul; Anderson, Karl E. (2012). "Trastornos de la biosíntesis del hemo". En Saudubray, Jean-Marie; van den Berghe, Georges; Walter, John H. (eds.). Enfermedades metabólicas congénitas: diagnóstico y tratamiento (5.ª ed.). Nueva York: Springer. págs. 521–532. ISBN 978-3-642-15719-6.
- ^ Ogun, Aminat S.; Joy, Neena V.; Valentine, Menogh (2022), "Bioquímica, síntesis de hemo", StatPearls , Treasure Island (FL): StatPearls Publishing, PMID 30726014 , consultado el 30 de octubre de 2022
- ^ "Pruebas para el diagnóstico de la porfiria". Fundación Americana de la Porfiria. Archivado desde el original el 20 de marzo de 2014. Consultado el 17 de mayo de 2014 .
- ^ Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ (2005). "Recomendaciones para el diagnóstico y tratamiento de las porfirias agudas". Ann. Intern. Med . 142 (6): 439–50. doi :10.7326/0003-4819-142-6-200503150-00010. PMID 15767622. S2CID 36122555.
- ^ Berkó G, Durkó I (19 de diciembre de 1971). "[Modificación del método Mauzerall-Granick para la determinación del ácido aminolevulínico urinario]". Orvosi Hetilap (en húngaro). 112 (51): 3085–6. PMID 5136653.
- ^ Woods, JS (1995). "Metabolismo de la porfirina como indicador de exposición y toxicidad a metales". En Goyer, RA; Cherian, MG (eds.). Toxicología de metales, aspectos bioquímicos . Vol. 115. Berlín: Springer. págs. 19–52, Capítulo 2.
- ^ Di Pierro, Elena; De Canio, Michele; Mercadante, Rosa; Savino, María; Granata, Francesca; Tavazzi, Darío; Nicolli, Anna María; Trevisan, Andrea; Marchini, Stefano; Fustinoni, Silvia (26 de julio de 2021). "Diagnóstico de laboratorio de porfiria". Diagnóstico . 11 (8): 1343. doi : 10.3390/diagnostics11081343 . ISSN 2075-4418. PMC 8391404 . PMID 34441276.
- ^ patient.co.uk. «Porfirias: tratamiento inmediato». Archivado desde el original el 20 de octubre de 2016. Consultado el 19 de octubre de 2016 .
- ^ Asociación Médica Británica , Sociedad Farmacéutica Real de Gran Bretaña (marzo de 2009). "9.8.2: Porfirias agudas". Formulario Nacional Británico (BNF 57) . Reino Unido: BMJ Group y RPS Publishing. p. 549. ISBN 978-0-85369-845-6.
- ^ Fundación Americana de Porfiria (2010). "Panhematin for Acute Porphyria". Archivado desde el original el 25 de agosto de 2010. Consultado el 5 de agosto de 2010 .
- ^ Cherem JH, Malagon J, Nellen H (2005). "Cimetidina y porfiria intermitente aguda". Ann. Intern. Med . 143 (9): 694–5. doi : 10.7326/0003-4819-143-9-200511010-00023 . PMID: 16263899.
- ^ "Porfiria - Diagnóstico y tratamiento - Mayo Clinic" www.mayoclinic.org . Consultado el 6 de diciembre de 2018 .
- ^ Birgisdottir BT, Asgeirsson H, Arnardottir S, Jonsson JJ, Vidarsson B (2010). "[Dolor abdominal agudo causado por porfiria intermitente aguda: informe de caso y revisión de la literatura]". Laeknabladid (en islandés). 96 (6): 413–18. PMID 20519771.
- ^ ab Tsao YC, Niu DM, Chen JT, Lin CY, Lin YY, Liao KK (2010). "La gabapentina reduce el dolor neurovisceral de la porfiria". Acta Neurol Taiwan . 19 (2): 112–5. PMID 20714961.
- ^ Cherian A, Thomas SV (2009). "Estado epiléptico". Ann Indian Acad Neurol . 12 (3): 140–53. doi : 10.4103/0972-2327.56312 . PMC 2824929 . PMID 20174493.
- ^ Murray ED, Buttner N, Price BH. (2012) Depresión y psicosis en la práctica neurológica. En: Neurología en la práctica clínica, sexta edición. Bradley WG, Daroff RB, Fenichel GM, Jankovic J (eds.) Butterworth Heinemann. 12 de abril de 2012. ISBN 978-1437704341
- ^ "Anticonceptivos hormonales y porfiria". Novedades sobre porfiria . Septiembre de 2020.
- ^ "La FDA aprueba el primer tratamiento para una enfermedad rara hereditaria". Administración de Alimentos y Medicamentos de Estados Unidos (FDA) (Comunicado de prensa). 20 de noviembre de 2019. Archivado desde el original el 21 de noviembre de 2019 . Consultado el 20 de noviembre de 2019 .
 Este artículo incorpora texto de esta fuente, que se encuentra en el dominio público .
Este artículo incorpora texto de esta fuente, que se encuentra en el dominio público . - ^ "La FDA aprueba el givosiran para la porfiria hepática aguda". Administración de Alimentos y Medicamentos de Estados Unidos (FDA) (Comunicado de prensa). 20 de noviembre de 2019. Archivado desde el original el 21 de noviembre de 2019 . Consultado el 20 de noviembre de 2019 .Este artículo incorpora texto de esta fuente, que se encuentra en el dominio público .
- ^ Martin A Crook. 2006. Química clínica y medicina metabólica. Séptima edición. Hodder Arnold. ISBN 0-340-90616-2
- ^ Faraci M, Morreale G, Boeri E, Lanino E, Dallorso S, Dini G, Scuderi F, Cohen A, Cappelli B (2008). "TCMH no relacionado en un adolescente afectado por porfiria eritropoyética congénita". Trasplante de Pediatría . 12 (1): 117–120. doi : 10.1111/j.1399-3046.2007.00842.x . PMID 18186900. S2CID 20789520.
- ^ «Registro comunitario de medicamentos de uso humano». ec.europa.eu . Comisión Europea. Archivado desde el original el 24 de diciembre de 2014 . Consultado el 24 de diciembre de 2014 .
- ^ Balwani, Manisha; Bonkovsky, Herbert L.; Levy, Cynthia; Anderson, Karl E.; Bissell, D. Montgomery; Parker, Charles; Takahashi, Fumihiro; Desnick, Robert J.; Belongie, Kirstine (13 de abril de 2023). "Dersimelagon en protoporfirias eritropoyéticas". New England Journal of Medicine . 388 (15): 1376–1385. doi :10.1056/NEJMoa2208754. ISSN 0028-4793. PMID 37043653. S2CID 258110676.
- ^ eMedicine > Porfiria cutánea Archivado el 5 de diciembre de 2010 en Wayback Machine. Autores: Vikramjit S Kanwar, Thomas G DeLoughery, Richard E Frye y Darius J Adams. Actualizado: 27 de julio de 2010.
- ^ Genetics Home Reference > Porfiria Archivado el 5 de agosto de 2010 en Wayback Machine Revisado en julio de 2009
- ^ Hoppe-Seyler F (1871). "Das Hämatin". Tubinger Med-Chem Untersuch . 4 : 523–33.
- ^ Nick Lane (16 de diciembre de 2002). «Born to the purple: the story of porphyria». Scientific American . Archivado desde el original el 11 de octubre de 2007. Consultado el 5 de agosto de 2008 .
- ^ Stokvis BJ. "Más de dos zeldzame kleurstoffen en orina van zieken". Nederl Tijdschr Geneeskd (en holandés). 2 : 409–417.Reimpreso en Stokvis BJ (diciembre de 1989). "Más de dos zeldzame kleurstoffen en orina van zieken". Ned Tijdschr Geneeskd (en holandés). 133 (51): 2562–70. PMID 2689889.
- ^ Denver, Joness. "Una enciclopedia de medicina oscura". Publicado por University Books, Inc., 1959.
- ^ Vampiros americanos: fans, víctimas, practicantes , Norine Dresser, WW Norton & Company, 1989.
- ^ "¿Los vampiros padecían la enfermedad llamada porfiria... o no?" Archivado el 5 de febrero de 2013 en Wayback Machine. The Straight Dope , 7 de mayo de 1999.
- ^ Cox, Ann M. (1995). "Porfiria y vampirismo: otro mito en ciernes". Revista Médica de Postgrado . 71 (841): 643–644. doi :10.1136/pgmj.71.841.643-a. PMC 2398345 . PMID 7494765.
- ^ Macalpine I, Hunter R (enero de 1966). "La 'insanidad' del rey Jorge III: un caso clásico de porfiria". Br Med J . 1 (5479): 65–71. doi :10.1136/bmj.1.5479.65. PMC 1843211 . PMID 5323262.
- ^ Macalpine I, Hunter R, Rimington C (enero de 1968). "Porfiria en las casas reales de Estuardo, Hannover y Prusia. Un estudio de seguimiento de la enfermedad de Jorge III". Br Med J . 1 (5583): 7–18. doi :10.1136/bmj.1.5583.7. PMC 1984936 . PMID 4866084.
- ^ Warren, Martin; Rh̲l, John CG; Hunt, David C. (1998). El secreto púrpura: genes, "locura" y las casas reales de Europa . Londres: Bantam Books . ISBN 978-0-593-04148-2.
- ^ Los autores demostraron una única mutación puntual en el gen PPOX, pero ninguna que haya sido asociada con la enfermedad.
- ^ Cox TM, Jack N, Lofthouse S, Watling J, Haines J, Warren MJ (2005). "El rey Jorge III y la porfiria: una hipótesis elemental y una investigación". The Lancet . 366 (9482): 332–335. doi :10.1016/S0140-6736(05)66991-7. PMID 16039338. S2CID 13109527.
- ^ Peters TJ, Wilkinson D (2010). "El rey Jorge III y la porfiria: una reexaminación clínica de la evidencia histórica". Historia de la psiquiatría . 21 (81 Pt 1): 3–19. doi :10.1177/0957154X09102616. PMID 21877427. S2CID 22391207.
- ^ Roberts, Andrew (9 de noviembre de 2021). "El último rey de Estados Unidos: el reinado incomprendido de Jorge III". Viking – vía Amazon.
- ^ Smith, Martin; Smith, Alison (21 de diciembre de 2009). Tetrapirroles: nacimiento, vida y muerte. Springer. ISBN 9780387785189Archivado desde el original el 4 de julio de 2014 . Consultado el 9 de abril de 2014 .
- ^ Röhl, John (25 de junio de 1999). «La bomba de tiempo tóxica de la familia real». Boletín: The University of Sussex Newsletter . Archivado desde el original el 15 de septiembre de 2013. Consultado el 18 de agosto de 2013 .
- ^ Roberts, Jenifer (2009). La locura de la reina María [ La extraordinaria vida de María I de Portugal ]. Templeton Press. ISBN 978-0954558918.
- ^ "BBC News - Reino Unido - Charles visita la casa de un "pariente" Drácula". Archivado desde el original el 24 de agosto de 2013.
- ^ Loftus LS, Arnold WN (1991). "La enfermedad de Vincent van Gogh: ¿porfiria intermitente aguda?". BMJ . 303 (6817): 1589–1591. doi :10.1136/bmj.303.6817.1589. PMC 1676250 . PMID 1773180.
- ^ Ranpura, Dhruv; Dhyey, Khavdu (2009) [1989]. La medicina de un hombre: una autobiografía del profesor Archie Cochrane . India: HarperCollins . ISBN 978-0-9540884-3-9.
- ^ Allende, Isabel (1995). Paula . Nueva York: HarperCollins. ISBN. 978-0-06-017253-4.
Enlaces externos
- Base de datos de medicamentos para la porfiria aguda: base de datos completa sobre la porfirinogenicidad de los medicamentos
- Página de Orphanet sobre la enfermedad de la porfiria