Amilosis
| Amilosis | |
|---|---|
 | |
| Los síntomas de la amiloidosis suelen ser vagos y requieren de distintos especialistas médicos para su diagnóstico. Los síntomas reveladores pueden incluir agrandamiento de la lengua ( macroglosia ) o hematomas alrededor de los ojos ( púrpura ) [1] | |
| Especialidad | Medicina interna |
| Síntomas | Sensación de cansancio, pérdida de peso, hinchazón de las piernas, dificultad para respirar, sangrado, sensación de mareo al estar de pie [2] |
| Inicio habitual | 55–65 años [2] |
| Causas | Genético o adquirido [3] |
| Método de diagnóstico | Biopsia de tejido [2] |
| Tratamiento | Tratamiento de apoyo , dirigido a la causa subyacente, diálisis , trasplante de órganos [3] |
| Pronóstico | Mejoró con el tratamiento [3] |
| Frecuencia | 3–13 por millón por año (amiloidosis AL) [2] |
| Fallecidos | 1 por cada 1.000 personas (mundo desarrollado) [3] |
La amiloidosis es un grupo de enfermedades en las que se acumulan proteínas anormales , conocidas como fibrillas amiloides , en el tejido. [4] Hay varios signos y síntomas no específicos y vagos asociados con la amiloidosis. [5] Estos incluyen fatiga, edema periférico , pérdida de peso , dificultad para respirar , palpitaciones y sensación de desmayo al estar de pie . [5] En la amiloidosis AL, los indicadores específicos pueden incluir agrandamiento de la lengua y púrpura periorbitaria . [5] En la amiloidosis ATTR de tipo salvaje, los síntomas no cardíacos incluyen: síndrome del túnel carpiano bilateral , estenosis espinal lumbar , ruptura del tendón del bíceps , neuropatía de fibras pequeñas y disfunción autonómica . [5]
Existen alrededor de 36 tipos diferentes de amiloidosis, cada uno debido a un mal plegamiento de proteínas específico . [6] Dentro de estas 36 proteínas, 19 se agrupan en formas localizadas , 14 se agrupan como formas sistémicas y tres proteínas pueden identificarse como cualquiera de ellas. [6] Estas proteínas pueden volverse irregulares debido a efectos genéticos, así como a través de factores ambientales adquiridos . [6] Los cuatro tipos más comunes de amiloidosis sistémica son la cadena ligera (AL) , la inflamación ( AA ), la relacionada con la diálisis (Aβ 2 M) y la hereditaria y de la vejez ( ATTR y amiloide transtiretina de tipo salvaje [7] ). [2]
El diagnóstico puede sospecharse cuando se encuentran proteínas en la orina , hay agrandamiento de órganos o se encuentran problemas con múltiples nervios periféricos y no está claro por qué. [2] El diagnóstico se confirma mediante una biopsia de tejido . [2] Debido a la presentación variable, a menudo puede llevar algún tiempo llegar al diagnóstico. [3]
El tratamiento está orientado a disminuir la cantidad de la proteína involucrada. [2] Esto a veces se puede lograr determinando y tratando la causa subyacente. [2] La amiloidosis AL ocurre en aproximadamente 3 a 13 por millón de personas por año y la amiloidosis AA en aproximadamente dos por millón de personas por año. [2] La edad habitual de aparición de estos dos tipos es de 55 a 60 años. [2] Sin tratamiento, la esperanza de vida es de entre seis meses y cuatro años. [2] En el mundo desarrollado, aproximadamente una de cada 1000 muertes se debe a amiloidosis sistémica. [3] La amiloidosis ha sido descrita desde al menos 1639. [2]
Signos y síntomas
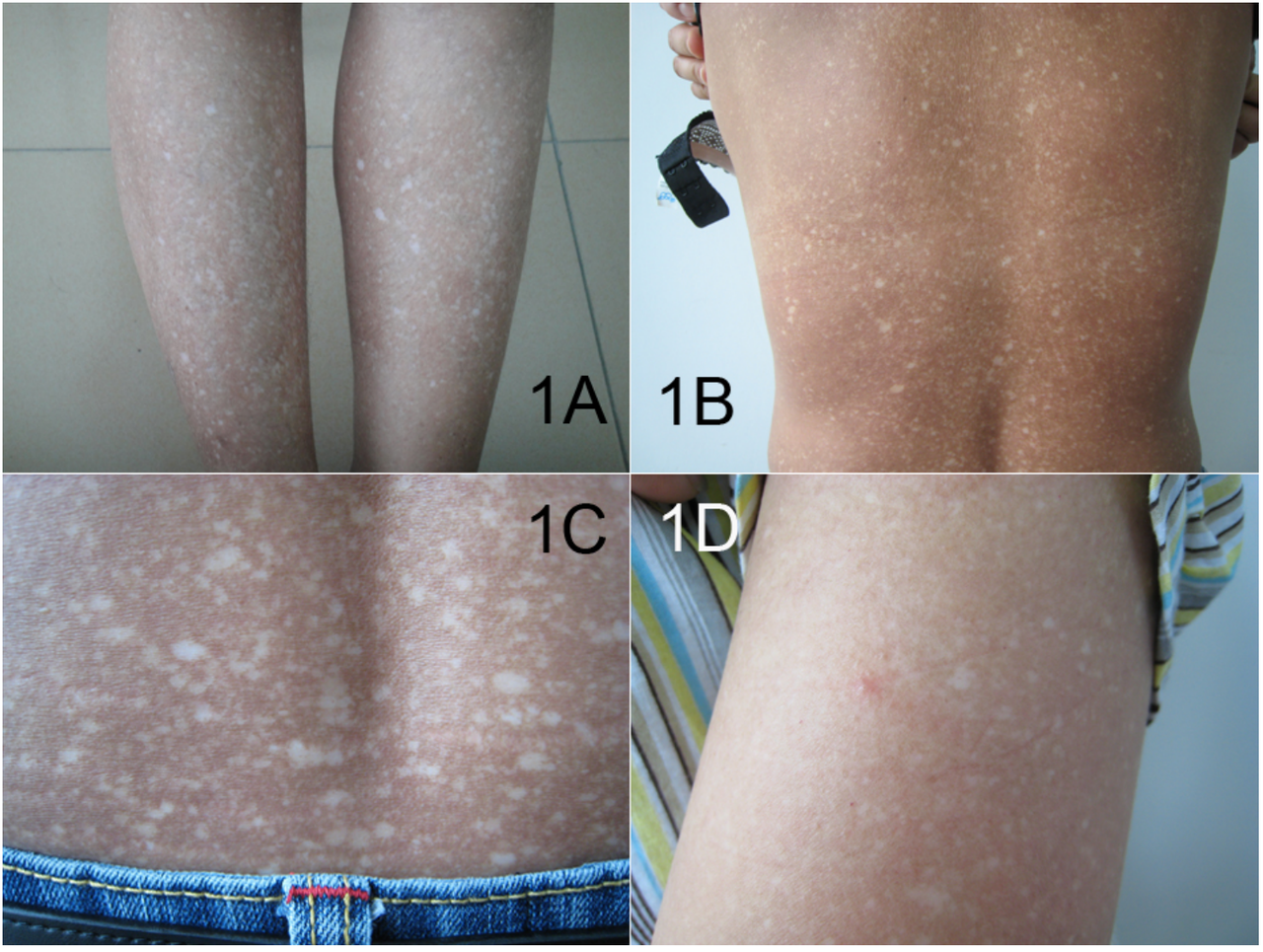
La presentación de la amiloidosis es amplia y depende del sitio de acumulación de amiloide. El riñón y el corazón son los órganos más comúnmente afectados.
Riñones
La deposición de amiloide en el riñón a menudo afecta los capilares glomerulares y las regiones mesangiales , lo que afecta la capacidad del órgano para filtrar y excretar desechos y retener proteínas plasmáticas . [8] Esto puede conducir a altos niveles de proteína en la orina ( proteinuria ) y síndrome nefrótico . [8] Varios tipos de amiloidosis, incluidos los tipos AL y AA, están asociados con el síndrome nefrótico . [9] Aproximadamente el 20% y el 40-60% de las personas con amiloidosis AL y AA respectivamente progresan a una enfermedad renal terminal que requiere diálisis . [9]
Corazón
La deposición de amiloide en el corazón puede causar insuficiencia cardíaca tanto diastólica como sistólica . Pueden presentarse cambios en el ECG , que muestran bajo voltaje y anomalías de la conducción como bloqueo auriculoventricular o disfunción del nódulo sinusal . [ cita médica requerida ] En la ecocardiografía , el corazón muestra un patrón de llenado restrictivo, con una función sistólica normal a levemente reducida. [ 10 ] La amiloidosis AA generalmente respeta el corazón. [ 11 ] La amiloidosis cardíaca puede presentarse con síntomas de insuficiencia cardíaca que incluyen dificultad para respirar, fatiga y edema. [ 12 ] A medida que progresa la amiloidosis cardíaca, la deposición de amiloide puede afectar la capacidad del corazón para bombear y llenarse de sangre, así como su capacidad para mantener el ritmo normal, lo que conduce al empeoramiento de la función cardíaca y al deterioro de la calidad de vida de las personas. [ 12 ]
Sistema nervioso
Las personas con amiloidosis pueden tener afectación del sistema nervioso central, [13] junto con afectación periférica que causa neuropatías sensitivas y autónomas. La neuropatía sensitiva se desarrolla en un patrón simétrico y progresa de manera distal a proximal. La neuropatía autónoma puede presentarse como hipotensión ortostática , pero puede manifestarse de manera más gradual con síntomas gastrointestinales inespecíficos como estreñimiento, náuseas o saciedad temprana. [10] La amiloidosis del sistema nervioso central puede tener presentaciones más graves y sistémicas que pueden incluir arritmias potencialmente mortales, insuficiencia cardíaca, desnutrición, infección o muerte. [14]
La presentación neuropática puede depender de la etiología de la amiloidosis. [14] Las personas con amiloidosis pueden experimentar disfunción en varios sistemas orgánicos dependiendo de la ubicación y el grado de afectación del sistema nervioso. [8] Por ejemplo, la neuropatía periférica puede causar disfunción eréctil, incontinencia y estreñimiento, disfunción pupilar y pérdida sensorial dependiendo de la distribución de la amiloidosis a lo largo de diferentes nervios periféricos. [14]
Órganos gastrointestinales y accesorios
La acumulación de proteínas amiloides en el sistema gastrointestinal puede ser causada por una amplia gama de trastornos amiloides y tiene diferentes presentaciones dependiendo del grado de afectación del órgano. [15] Los síntomas potenciales incluyen pérdida de peso, diarrea, dolor abdominal, acidez estomacal (reflujo gastrointestinal) y sangrado gastrointestinal. [15] La amiloidosis también puede afectar los órganos digestivos accesorios, incluido el hígado, y puede presentarse con ictericia, heces grasas, anorexia, acumulación de líquido en el abdomen y agrandamiento del bazo. [15]
La acumulación de proteínas amiloides en el hígado puede provocar elevaciones de las aminotransferasas séricas y la fosfatasa alcalina , dos biomarcadores de lesión hepática, que se observan en aproximadamente un tercio de las personas. [11] El agrandamiento del hígado es común. En contraste, el agrandamiento del bazo es raro y ocurre en el 5% de las personas. [10] La disfunción esplénica, que conduce a la presencia de cuerpos de Howell-Jolly en el frotis de sangre, ocurre en el 24% de las personas con amiloidosis. [10] La malabsorción se observa en el 8,5% de la amiloidosis AL y en el 2,4% de la amiloidosis AA. Un mecanismo sugerido para la malabsorción observada es que los depósitos de amiloide en las puntas de las vellosidades intestinales (proyecciones en forma de dedos que aumentan el área intestinal disponible para la absorción de alimentos), comienzan a erosionar la funcionalidad de las vellosidades, presentando un cuadro similar al esprú . [11]
Glándulas
Tanto la glándula tiroides como las glándulas suprarrenales pueden estar infiltradas. Se estima que entre el 10 y el 20 % de las personas con amiloidosis tienen hipotiroidismo . La infiltración suprarrenal puede ser más difícil de detectar dado que sus síntomas de hipotensión ortostática y baja concentración de sodio en sangre pueden atribuirse a neuropatía autonómica e insuficiencia cardíaca. [10]
"Los depósitos de amiloide se producen en el páncreas de las personas que también tienen diabetes mellitus , aunque no se sabe si esto es funcionalmente importante. El componente principal del amiloide pancreático es un péptido de 37 aminoácidos conocido como polipéptido amiloide de los islotes o 'amilina'. Este se almacena con la insulina en gránulos secretores en las células [beta] y se secreta conjuntamente con la insulina". (Rang and Dale's Pharmacology, 2015.) [ cita requerida ]
Sistema musculoesquelético
Las proteínas amiloides se depositan con mayor frecuencia en el interior de la rodilla, seguidas de las manos, las muñecas, los codos, la cadera y el tobillo, lo que provoca dolor articular. [16] En los hombres de edad avanzada (>80 años), existe un riesgo significativo de depósito de amiloide transtiretina de tipo salvaje en el tejido sinovial de la articulación de la rodilla, pero predominantemente en la vejez, el depósito de transtiretina de tipo salvaje se observa en los ventrículos cardíacos. Se han encontrado depósitos de ATTR en el ligamento amarillo de pacientes que se sometieron a cirugía por estenosis espinal lumbar . [17]
En la amiloidosis beta 2-microglobulina, los hombres tienen un alto riesgo de contraer el síndrome del túnel carpiano . [18] La amiloidosis Aβ2MG (amiloidosis asociada a hemodiálisis) tiende a depositarse en el tejido sinovial, causando inflamación crónica del tejido sinovial en la rodilla, la cadera, el hombro y las articulaciones interfalángicas. [18] El depósito de cadenas ligeras amiloides en la articulación del hombro causa hombros agrandados, también conocido como " signo de la hombrera ". [18] Los depósitos de cadenas ligeras amiloides también pueden causar poliartritis simétrica bilateral. [18]
El depósito de proteínas amiloides en la médula ósea sin causar discrasias de células plasmáticas se denomina amiloidoma. Se encuentra comúnmente en las vértebras cervicales, lumbares y sacras. Los afectados pueden presentar dolor óseo debido a la lisis ósea, paraparesia lumbar y una variedad de síntomas neurológicos. Las fracturas vertebrales también son comunes. [18]
Ojos
Una rara manifestación es la púrpura amiloide , una susceptibilidad a sangrado con hematomas alrededor de los ojos, denominada "ojos de mapache". La púrpura amiloide es causada por la deposición de amiloide en los vasos sanguíneos y la actividad reducida de la trombina y el factor X , dos proteínas de coagulación que pierden su función después de unirse con amiloide. [10]
Cavidad bucal
Los depósitos de amiloide en el tejido pueden causar agrandamiento de las estructuras. El veinte por ciento de las personas con amiloidosis AL tienen la lengua agrandada , lo que puede provocar apnea obstructiva del sueño , dificultad para tragar y alteración del gusto. [11] El agrandamiento de la lengua no ocurre en la amiloidosis ATTR o AA. [10] El depósito de amiloide en la garganta puede causar ronquera. [10]
Patogenesia
Las amiloidosis pueden considerarse enfermedades de plegamiento incorrecto de proteínas . [19] [20] La gran mayoría de las proteínas que se ha descubierto que forman depósitos amiloides son proteínas secretadas , por lo que el plegamiento incorrecto y la formación de amiloide se producen fuera de las células, en el espacio extracelular . [19] De las 37 proteínas identificadas hasta ahora como vulnerables a la formación de amiloide, solo cuatro son citosólicas . [19] La mayoría de las proteínas formadoras de amiloide son relativamente pequeñas, pero por lo demás actualmente no hay evidencia de similitudes estructurales o funcionales entre las proteínas que se sabe que forman amiloides asociados a la enfermedad. [19] Un tercio de la enfermedad amiloide es hereditaria, en cuyo caso normalmente hay una edad temprana de aparición. [19] La mitad de las enfermedades relacionadas con amiloide son esporádicas y tienen una edad tardía de aparición; en estos casos, la agregación de proteínas puede estar asociada con la disminución relacionada con el envejecimiento en la regulación de las proteínas. Algunos tratamientos médicos están asociados con la enfermedad amiloide, pero esto es poco común. [19]
Las proteínas formadoras de amiloide se agregan en formas fibrilares distintivas con una estructura de lámina beta . [19] [20] La forma de lámina beta del amiloide es resistente a la proteólisis , lo que significa que no se puede degradar ni descomponer. [5] Como resultado, el amiloide se deposita en el espacio extracelular del cuerpo. [5] Se cree que el proceso de formación de fibrillas amiloides tiene formas oligoméricas intermedias . Tanto los oligómeros como las fibrillas amiloides pueden ser tóxicos para las células y pueden interferir con el funcionamiento adecuado de los órganos. [21] La importancia relativa de las diferentes especies de agregación puede depender de la proteína involucrada y del sistema orgánico afectado. [20]
Diagnóstico
El diagnóstico de la amiloidosis generalmente requiere una biopsia de tejido. [2] La biopsia se evalúa para detectar la presencia de depósitos amiloides característicos. El tejido se trata con varias tinciones . La tinción más útil para el diagnóstico de amiloide es el rojo Congo , que, combinado con luz polarizada , hace que las proteínas amiloides aparezcan de color verde manzana en el microscopio . También se puede utilizar la tinción con tioflavina T. [22] También se utilizan varias técnicas de diagnóstico por imágenes, como la gammagrafía PYP de medicina nuclear, la gammagrafía DPD o la gammagrafía SAP . [23]
Se puede realizar una biopsia de una muestra de tejido o se puede obtener directamente del órgano interno afectado, pero el sitio de primera línea de la biopsia es la grasa abdominal subcutánea , conocida como "biopsia de la almohadilla grasa", debido a su facilidad de adquisición. [24] [25] Una biopsia de grasa abdominal no es completamente sensible y puede dar lugar a falsos negativos , lo que significa que un resultado negativo no excluye el diagnóstico de amiloidosis. [24] [25] Sin embargo, la biopsia directa del órgano afectado puede seguir siendo innecesaria, ya que también se pueden utilizar otros métodos de biopsia menos invasivos, incluida la biopsia de la mucosa rectal, de la glándula salival, del labio o de la médula ósea, que pueden lograr un diagnóstico en hasta el 85% de las personas. [24]
En el depósito de amiloide en las articulaciones, habrá una señal disminuida en las imágenes de resonancia magnética ponderadas en T1 y T2 . [16] En el amiloidoma, habrá una señal baja en T1 con la inyección de gadolinio y una señal baja en T2. [18]
El tipo de proteína amiloide se puede determinar de varias maneras: la detección de proteínas anormales en el torrente sanguíneo (en la electroforesis de proteínas o determinación de la cadena ligera); la unión de anticuerpos particulares al amiloide encontrado en el tejido (inmunohistoquímica); o la extracción de la proteína e identificación de sus aminoácidos individuales . [22] La inmunohistoquímica puede identificar la amiloidosis AA la mayoría de las veces, pero puede pasar por alto muchos casos de amiloidosis AL. [11] La microdisección láser con espectrometría de masas es el método más confiable para identificar las diferentes formas de amiloidosis. [26]
La AL se consideraba anteriormente la forma más común de amiloidosis, y el diagnóstico a menudo comienza con la búsqueda de discrasia de células plasmáticas , células B de memoria que producen inmunoglobulinas aberrantes o porciones de inmunoglobulinas. La electroforesis de inmunofijación de orina o suero es positiva en el 90% de las personas con amiloidosis AL. [10] La electroforesis de inmunofijación es más sensible que la electroforesis regular, pero puede no estar disponible en todos los centros. Alternativamente, se puede buscar la tinción inmunohistoquímica de una biopsia de médula ósea en busca de células plasmáticas dominantes en personas con una alta sospecha clínica de amiloidosis AL pero electroforesis negativa. [10]
Actualmente, se considera que la ATTR es la forma más común de amiloidosis. Puede estar relacionada con la edad en el caso de la ATTR de tipo salvaje (ATTRv) o ser una amiloidosis familiar asociada a la transtiretina; se sospecha en personas con antecedentes familiares de neuropatías idiopáticas o insuficiencia cardíaca que no presentan evidencia de discrasias de células plasmáticas. La ATTR se puede identificar mediante el uso de enfoque isoeléctrico que separa las formas mutadas de la transtiretina. Los hallazgos se pueden corroborar mediante pruebas genéticas para buscar mutaciones específicas conocidas en la transtiretina que predisponen a la amiloidosis. [10]
La AA se sospecha clínicamente en personas con infecciones o enfermedades inflamatorias de larga duración. La AA se puede identificar mediante tinción inmunohistoquímica. [10]
- Intestino delgado y duodeno con depósito de amiloide Rojo Congo 10X
- Amiloidosis, calcificación distrófica
- Intestino delgado y duodeno con depósito de amiloide 20X
- Amiloidosis, ganglio, rojo Congo
- Amiloidosis, vasos sanguíneos, H&E
- Amiloidosis, ganglio linfático, H&E
- Amiloidosis, ganglio linfático, polarizador
- Micrografía que muestra el depósito de amiloide (material rojo esponjoso) en el corazón ( amiloidosis cardíaca ). Tinción de rojo Congo .








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clasificación
Los sistemas de clasificación históricos se basaban en factores clínicos. Hasta principios de la década de 1970, predominaba la idea de una única sustancia amiloide. Se propusieron varios sistemas de clasificación descriptiva basados en la distribución orgánica de los depósitos de amiloide y los hallazgos clínicos. La mayoría de los sistemas de clasificación incluían la amiloidosis primaria (es decir, idiopática ), en la que no se identificaba ninguna afección clínica asociada, y la amiloidosis secundaria (es decir, secundaria a afecciones inflamatorias crónicas). Algunos sistemas de clasificación incluían la amiloidosis asociada al mieloma, familiar y localizada. [ cita requerida ]
La era moderna de la clasificación de la amiloidosis comenzó a fines de la década de 1960 con el desarrollo de métodos para hacer solubles las fibrillas de amiloide. Estos métodos permitieron a los científicos estudiar las propiedades químicas de los amiloides. [ cita médica requerida ] Los términos descriptivos como amiloidosis primaria, amiloidosis secundaria y otros (por ejemplo, amiloidosis senil), que no se basan en la causa, brindan poca información útil y ya no se recomiendan.
La clasificación moderna de la enfermedad amiloide tiende a utilizar una abreviatura de la proteína que produce la mayoría de los depósitos, precedida por la letra A. Por ejemplo, la amiloidosis causada por la transtiretina se denomina "ATTR". [ cita médica necesaria ] Los patrones de deposición varían entre las personas, pero casi siempre están compuestos por una sola proteína amiloidogénica. La deposición puede ser sistémica (que afecta a muchos sistemas orgánicos diferentes) o específica de un órgano. Muchas amiloidosis son hereditarias , debido a mutaciones en la proteína precursora. [ cita médica necesaria ]
Otras formas se deben a diferentes enfermedades que causan una producción de proteínas sobreabundante o anormal, como la sobreproducción de cadenas ligeras de inmunoglobulina (denominada amiloidosis AL ) o la sobreproducción continua de proteínas de fase aguda en la inflamación crónica (que puede conducir a la amiloidosis AA ). [ cita médica necesaria ]
Hasta el momento se han identificado alrededor de 60 proteínas amiloides. [27] De ellas, al menos 36 se han asociado con una enfermedad humana. [28]
Todas las proteínas de fibrillas amiloides comienzan con la letra "A" seguida del sufijo de la proteína (y cualquier especificación aplicable). Vea a continuación una lista de proteínas de fibrillas amiloides que se han encontrado en humanos: [29]
| Proteína fibrilar | Proteína precursora | Órganos diana | Sistémico y/o localizado | Adquirida o hereditaria |
|---|---|---|---|---|
| Alabama | Cadena ligera de inmunoglobulina | Todos los órganos, generalmente excepto el SNC | Yo, Yo | A, H |
| Ah | Cadena pesada de inmunoglobulina | Todos los órganos excepto el SNC | Yo, Yo | A |
| Automóvil club británico | (Apo) amiloide sérico A | Todos los órganos excepto el SNC | S | A |
| ATRIBUTO | Transtiretina , tipo salvaje Transtiretina , variantes | Corazón principalmente en varones, pulmón, ligamentos, tenosinovio PNS, ANS, corazón, ojo, leptomeninges | S S | A yo |
| Aβ2M | β2-microglobulina , tipo salvaje β2-microglobulina , variantes | Sistema musculoesquelético Respuesta | S S | A yo |
| AapoAI | Apolipoproteína AI , variantes | Corazón, hígado, riñón, SNP, testículos, laringe (C variantes terminales), piel (variantes terminales C) | S | yo |
| AapoAII | Apolipoproteína A II , variantes | Riñón | S | yo |
| AapoAIV | Apolipoproteína A IV, tipo salvaje | Médula renal y sistémica | S | A |
| AapoCII | Apolipoproteína C II, variantes | Riñón | S | yo |
| ApoCIII | Apolipoproteína C III, variantes | Riñón | S | yo |
| Agel | Gelsolina, variantes | Riñón, SNP, córnea | S | yo |
| ALys | Lisozima, variantes | Riñón | S | yo |
| ALECT2 | Factor quimiotáctico leucocitario-2 | Riñón, principalmente | S | A |
| Fibrilación auricular | Fibrinógeno a, variantes | Riñón, principalmente | S | yo |
| ACys | Variantes de la cistatina C | SNC , SNP, piel | S | yo |
| Abri | ABriPP, variantes | Sistema nervioso central | S | yo |
| Adán b | ADanPP, variantes | Sistema nervioso central | yo | yo |
| Aβ | Precursor de la proteína Aβ, tipo salvaje Precursor de la proteína Aβ, variante | Sistema nervioso central | yo yo | A yo |
| AαSiN | α-sinucleína | Sistema nervioso central | yo | A |
| Un Au | Tauro | Sistema nervioso central | yo | A |
| APrP | Proteína priónica, tipo salvaje Variantes de la proteína priónica Variante de la proteína priónica | Enfermedad de Creutzfeldt-Jakob (ECJ), insomnio mortal CJD, síndrome GSS, insomnio letal PNS | yo yo S | A yo yo |
| Acal | (Pro)calcitonina | Tumores tiroideos de células C Riñón | yo S | A A |
| Aplicación AI | Polipéptido amiloide c de los islotes | Islotes de Langerhans, insulinomas | yo | A |
| Asociación Estadounidense de Fútbol Americano | Factor natriurético auricular | Aurículas cardíacas | yo | A |
| Apro | Prolactina | Prolactinomas hipofisarios, envejecimiento de la hipófisis | yo | A |
| AIn | Insulina | Inyección local iatrogénica | yo | A |
| ASPC- d | Proteína surfactante pulmonar | Pulmón | yo | A |
| ACor | Corneodesmosina | Epitelios cornificados, folículos pilosos | yo | A |
| Amado | Lactadherina | Aorta senil, media | yo | A |
| Aker (Aker) | Queratoepitelina | Córnea, hereditaria | yo | A |
| ALac | Lactoferrina | Córnea | yo | A |
| AOAAP | Proteína odontogénica asociada a ameloblastos | Tumores odontogénicos | yo | A |
| ASem1 | Semenogelina 1 | Vesícula seminal | yo | A |
| AEnf | Enfurvitida | Iatrogénico | yo | A |
| ACatK y | Catepsina K | Asociado a tumores | yo | A |
| AEFEMP1 y | Fibulina extracelular que contiene EGF proteína de matriz 1 (EFEMP1) | Venas porta asociadas al envejecimiento | yo | A |
Alternativa
Un método de clasificación clínica más antiguo se refiere a las amiloidosis como sistémicas o localizadas:
- Las amiloidosis sistémicas afectan a más de un órgano o sistema corporal. Algunos ejemplos son AL, AA y Aβ2m. [30]
- Las amiloidosis localizadas afectan solo a un órgano o tipo de tejido corporal. Algunos ejemplos son Aβ , IAPP , factor natriurético auricular (en la amiloidosis auricular aislada ) y calcitonina (en el carcinoma medular de tiroides ) [30]
Otra clasificación es primaria o secundaria. [ cita médica necesaria ]
- Las amiloidosis primarias surgen de una enfermedad con función alterada de las células inmunes, como el mieloma múltiple u otras discrasias de inmunocitos.
- Las amiloidosis secundarias (reactivas) se producen como complicación de alguna otra enfermedad inflamatoria crónica o destructora de tejidos. Algunos ejemplos son la amiloidosis sistémica reactiva y la amiloidosis cutánea secundaria . [30]
Además, en función de los tejidos en los que se deposita, se divide en mesenquimal (órganos derivados del mesodermo ) o parenquimal (órganos derivados del ectodermo o endodermo ). [ cita médica necesaria ]
Tratamiento
El tratamiento depende del tipo de amiloidosis presente. El tratamiento con dosis altas de melfalán , un agente quimioterapéutico , seguido de un trasplante de células madre ha demostrado ser prometedor en estudios preliminares y se recomienda para la amiloidosis AL en estadio I y II. [26] Sin embargo, solo el 20-25% de las personas son elegibles para el trasplante de células madre. El tratamiento de quimioterapia que incluye ciclofosfamida-bortezomib-dexametasona es actualmente la opción de tratamiento recomendada para las personas con amiloidosis AL que no son elegibles para el trasplante. [5]
En la amiloidosis AA, los síntomas pueden mejorar si se trata la afección subyacente. En las personas que tienen inflamación causada por la amiloidosis AA, se utilizan inhibidores del factor de necrosis tumoral (TNF) alfa, como infliximab y etanercept , durante una duración media de 20 meses. Si los inhibidores del TNF alfa no son eficaces, se pueden considerar los inhibidores de la interleucina-1 (p. ej., anakinra , canakinumab , rilonacept ) y los inhibidores de la interleucina-6 (p. ej., tocilizumab). [31]
El tratamiento de la amiloidosis ATTR dependerá de su clasificación como tipo salvaje o variante. [5] Ambos pueden tratarse con tafamidis , un agente oral de baja toxicidad que previene la desestabilización de la proteína correctamente plegada. [5] Los estudios demostraron que tafamidis redujo la mortalidad y la hospitalización por insuficiencia cardíaca . [5] Anteriormente, para la amiloidosis ATTR variante, el trasplante de hígado era el único tratamiento eficaz. [5] Las nuevas terapias incluyen diflunisal , inotersén y patisirán .
El diflunisal se une a la proteína TTR mutante mal plegada para evitar su acumulación, de la misma manera que lo hace el tafamidis. La evidencia de baja certeza indica que mitiga el empeoramiento de la neuropatía periférica y la discapacidad derivada de la progresión de la enfermedad. [32]
El inotersén bloquea la expresión génica de la TTR tanto de tipo salvaje como mutante, lo que reduce el precursor amiloide. La evidencia de certeza moderada sugiere que mitiga el empeoramiento de la neuropatía periférica. La eficacia y seguridad a largo plazo del uso de inotersén en personas con amiloidosis relacionada con la TTR mutante aún se está evaluando en un ensayo clínico de fase III a partir de 2021. Tanto el diflunisal como el inotersén también pueden mitigar las disminuciones en la calidad de vida, aunque la evidencia de este efecto no es clara. [32] Para las personas con ATTR cardíaca, el efecto del uso de inotersén no es concluyente y requiere más investigación. [33] En 2018, la Agencia Europea de Medicamentos aprobó el inotersén para tratar la polineuropatía en adultos con amiloidosis hereditaria por transtiretina. [34] Desde entonces, ha sido aprobado para su uso en Canadá, la Unión Europea y en los EE. UU. [35]
El patisirán funciona de manera similar al inotersén. La evidencia de certeza moderada sugiere que el patisirán mitiga el empeoramiento de la neuropatía periférica y la discapacidad por la progresión de la enfermedad. Además, la evidencia de certeza baja sugiere que el patisirán mitiga las disminuciones en la calidad de vida y reduce levemente la tasa de eventos adversos en comparación con el placebo. No hay evidencia de un efecto sobre la tasa de mortalidad. [32] Una revisión de los datos iniciales del uso de patisirán en personas con ATTR cardíaca variante sugiere que puede reducir la mortalidad y la hospitalización, sin embargo, esto todavía se está investigando y requiere más investigación. [33] En 2018, el NICE no recomendó el patisirán en el Reino Unido para la amiloidosis hereditaria relacionada con la transtiretina. [36] Sin embargo, a partir de julio de 2019 se están realizando más revisiones. [37] Sin embargo, fue aprobado para este uso en los Estados Unidos. [38]
Todavía se están investigando los roles del inotersén y el patisirán en la amiloidosis ATTR cardíaca. [5]
En 2021, en un ensayo clínico que utilizó la técnica de edición genética CRISPR , varios participantes tuvieron una "caída del 80% al 96% en los niveles de TTR, igual o mejor que el promedio del 81%" de quienes recibieron patisirán. [39]
La Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) aprobó el vutrisiran en junio de 2022 para el tratamiento de la polineuropatía de la amiloidosis hereditaria mediada por transtiretina (hATTR) en adultos. [40]
Grupos de apoyo
Las personas afectadas por amiloidosis reciben apoyo de organizaciones como el Consorcio de Investigación de Amiloidosis, la Fundación de Amiloidosis, los Grupos de Apoyo a la Amiloidosis y la Red Australiana de Amiloidosis. [41] [42]
Pronóstico
El pronóstico varía según el tipo de amiloidosis y el sistema orgánico afectado. El pronóstico para la amiloidosis cardíaca AL no tratada es malo, con una supervivencia media de seis meses. [43] Más específicamente, la amiloidosis AL se puede clasificar como estadio I, II o III según biomarcadores cardíacos como Nt-proBNP y troponina cardíaca. [44] La supervivencia disminuye con el aumento del estadio, pero los avances recientes en los tratamientos han mejorado las tasas de supervivencia media para los estadios I, II y III, a 91,2, 60 y 7 meses respectivamente. [44]
Los resultados en una persona con amiloidosis AA dependen de la enfermedad subyacente, los órganos afectados y se correlacionan con la concentración de proteína amiloide A sérica. [5]
Las personas con ATTR, ATTR mutante y ATTR de tipo salvaje tienen un mejor pronóstico en comparación con las personas con AL y pueden sobrevivir durante más de una década. [10] [45] El tiempo de supervivencia no está asociado con el género o la edad, sin embargo, algunas medidas de función cardíaca reducida están asociadas con un tiempo de supervivencia más corto. [45]
Se determinó que la amiloidosis sistémica senil es la principal causa de muerte en el 70% de las personas mayores de 110 años a las que se les realizó una autopsia . [46] [47]
Epidemiología
La amiloidosis tiene una prevalencia combinada estimada de 30 por 100.000 personas y las tres formas más comunes son AL, ATTR y AA. [48] La edad media en el momento del diagnóstico es de 64 años. [11]
La AL tiene la incidencia más alta, aproximadamente 12 casos por millón de personas por año, y una prevalencia estimada de 30 000 a 45 000 casos en los EE. UU. y la Unión Europea. [48] [5]
La amiloidosis AA es la forma más común en los países en desarrollo y puede complicar infecciones de larga duración como tuberculosis , osteomielitis y bronquiectasias . La amiloidosis AA es causada por un aumento en la deposición extracelular de la proteína amiloide sérica A (SAA). Los niveles de proteína SAA pueden aumentar de manera directa e indirecta, a través de infecciones, inflamaciones y neoplasias malignas. [49] Las causas más comunes de amiloidosis AA en Occidente son la artritis reumatoide, la enfermedad inflamatoria intestinal, la psoriasis y la fiebre mediterránea familiar . [10]
Las personas sometidas a hemodiálisis a largo plazo (14 a 15 años) pueden desarrollar amiloidosis por acumulación de cadenas ligeras del complejo HLA 1 que normalmente se filtra por los riñones. [11]
La amiloidosis por transtiretina (ATTR) de tipo salvaje se encuentra en una cuarta parte de los ancianos en la autopsia. [50] La ATTR se encuentra en el 13-19 % de las personas que experimentan insuficiencia cardíaca con fracción de eyección preservada, lo que la convierte en una forma muy común de amiloidosis sistémica. [51]
Investigación
Los tratamientos para la neuropatía relacionada con ATTR incluyen oligonucleótidos específicos de TTR en forma de ARN interferente pequeño (patisirán) o inotersén antisentido , [52] el primero ha recibido recientemente la aprobación de la FDA. [53] Las investigaciones sobre tratamientos para la amiloidosis por ATTR han comparado el trasplante de hígado, medicamentos orales que estabilizan la proteína mal plegada (incluidos tafamidis y diflunisal) y agentes terapéuticos más nuevos que aún se están investigando (incluido patisirán). [54]
Según las investigaciones disponibles, el trasplante de hígado sigue siendo la opción de tratamiento más eficaz para la amiloidosis ATTR avanzada, los fármacos estabilizadores de proteínas pueden retardar la progresión de la enfermedad pero fueron insuficientes para justificar el retraso del trasplante de hígado, y los agentes más nuevos como el patisirán requieren estudios adicionales. [54]
Véase también
Referencias
- ^ "Concientización sobre la amiloidosis" (PDF) . amyloidaware.com . Grupos de apoyo para la amiloidosis. 1 de marzo de 2022 . Consultado el 15 de junio de 2024 .
- ^ abcdefghijklmn Hazenberg BP (mayo de 2013). "Amiloidosis: una descripción clínica" (PDF) . Clínicas de enfermedades reumáticas de Norteamérica . 39 (2): 323–345. doi :10.1016/j.rdc.2013.02.012. PMID 23597967. S2CID 215069282.
- ^ abcdef Pepys MB (2006). "Amiloidosis". Revista Anual de Medicina . 57 : 223–241. doi :10.1146/annurev.med.57.121304.131243. PMID 16409147.
- ^ "Amiloidosis AL". rarediseases.info.nih.gov . Centro de Información sobre Enfermedades Genéticas y Raras (GARD). Archivado desde el original el 24 de abril de 2017 . Consultado el 22 de abril de 2017 .
- ^ abcdefghijklmn Gertz MA, Dispenzieri A (julio de 2020). "Reconocimiento, pronóstico y tratamiento de la amiloidosis sistémica: una revisión sistemática". JAMA . 324 (1): 79–89. doi :10.1001/jama.2020.5493. PMID 32633805. S2CID 220385853.
- ^ abc Picken MM (2020). "La patología de la amiloidosis en la clasificación: una revisión". Acta Haematologica . 143 (4): 322–334. doi : 10.1159/000506696 . PMID 32392555. S2CID 218600304.
- ^ Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S, et al. (febrero de 2013). "Guía de amiloidosis hereditaria relacionada con la transtiretina para médicos". Orphanet Journal of Rare Diseases . 8 : 31. doi : 10.1186/1750-1172-8-31 . PMC 3584981 . PMID 23425518.
- ^ abc "Amiloidosis y enfermedad renal". Instituto Nacional de Diabetes y Enfermedades Digestivas y Renales . Departamento de Salud y Servicios Humanos de EE. UU. Archivado desde el original el 19 de noviembre de 2021. Consultado el 19 de noviembre de 2021 .
- ^ ab Lewis JB, Neilson EG (2018). "Enfermedades glomerulares". En Jameson J, Fauci AS, Kasper DL, Hauser SL, Longo DL, Loscalzo J (eds.). Principios de medicina interna de Harrison (20.ª ed.). McGraw Hill. Archivado desde el original el 29 de noviembre de 2021. Consultado el 29 de noviembre de 2021 .
- ^ abcdefghijklmn Falk RH, Comenzo RL, Skinner M (septiembre de 1997). "Las amiloidosis sistémicas". The New England Journal of Medicine . 337 (13): 898–909. doi :10.1056/NEJM199709253371306. PMID 9302305.
- ^ abcdefg Ebert EC, Nagar M (marzo de 2008). "Manifestaciones gastrointestinales de la amiloidosis". La Revista Estadounidense de Gastroenterología . 103 (3): 776–787. doi :10.1111/j.1572-0241.2007.01669.x. PMID 18076735. S2CID 25431033.
- ^ ab Kyriakou P, Mouselimis D, Tsarouchas A, Rigopoulos A, Bakogiannis C, Noutsias M, Vassilikos V (diciembre de 2018). "Diagnóstico de amiloidosis cardíaca: una revisión sistemática sobre el papel de las imágenes y los biomarcadores". Trastornos cardiovasculares del BMC . 18 (1): 221. doi : 10.1186/s12872-018-0952-8 . PMC 6278059 . PMID 30509186.
- ^ Soprano DR, Herbert J, Soprano KJ, Schon EA, Goodman DS. Demostración de ARNm de transtiretina en el cerebro y otros tejidos extrahepáticos en ratas. J Biol Chem 1985; 260 (21) 11793-11798
- ^ abc Kaku M, Berk JL (octubre de 2019). "Neuropatía asociada con amiloidosis sistémica". Seminarios en neurología . 39 (5): 578–588. doi :10.1055/s-0039-1688994. PMID 31639841. S2CID 204850185.
- ^ abc Rowe K, Pankow J, Nehme F, Salyers W (mayo de 2017). "Amiloidosis gastrointestinal: revisión de la literatura". Cureus . 9 (5): e1228. doi : 10.7759/cureus.1228 . PMC 5464793 . PMID 28611935.
- ^ ab Takahashi N, Glockner J, Howe BM, Hartman RP, Kawashima A (mayo de 2016). "Taxonomía y manifestaciones por imágenes de la amiloidosis sistémica". Radiologic Clinics of North America . 54 (3): 597–612. doi :10.1016/j.rcl.2015.12.012. PMID 27153791.
- ^ Eldhagen P, Berg S, Lund LH, Sörensson P, Suhr OB, Westermark P (junio de 2021). "Depósitos de amiloide de transtiretina en la estenosis espinal lumbar y evaluación de signos de amiloidosis sistémica". Revista de Medicina Interna . 289 (6): 895–905. doi :10.1111/joim.13222. ISSN 1365-2796. PMC 8248398 . PMID 33274477.
- ^ abcdef Nguyen TX, Naqvi A, Thompson TL, Wilson RH (primavera de 2018). "Manifestaciones musculoesqueléticas de la amiloidosis: una revisión centrada". Revista de avances en cirugía ortopédica . 27 (1): 1–5. PMID 29762107.
- ^ abcdefg Chiti F, Dobson CM (junio de 2017). "Plegamiento incorrecto de proteínas, formación de amiloide y enfermedades humanas: un resumen del progreso durante la última década". Revisión anual de bioquímica . 86 : 27–68. doi :10.1146/annurev-biochem-061516-045115. hdl : 2158/1117236 . PMID 28498720.
- ^ abc Merlini G, Seldin DC, Gertz MA (mayo de 2011). "Amiloidosis: patogénesis y nuevas opciones terapéuticas". Journal of Clinical Oncology . 29 (14): 1924–1933. doi :10.1200/JCO.2010.32.2271. PMC 3138545 . PMID 21483018.
- ^ Gertz MA, Rajkumar SV (2010). Amiloidosis . Totowa, Nueva Jersey: Humana. ISBN 978-1-60761-631-3.OCLC 654382006 .
- ^ ab Dember LM (diciembre de 2006). «Amiloidosis-associated renal disease» (Enfermedad renal asociada a amiloidosis). Journal of the American Society of Nephrology (Revista de la Sociedad Estadounidense de Nefrología ). 17 (12): 3458–3471. doi : 10.1681/ASN.2006050460 . PMID 17093068. Archivado desde el original el 5 de diciembre de 2011.
- ^ Sachchithanantham S, Wechalekar AD (2013). "Imágenes en amiloidosis sistémica". British Medical Bulletin . 107 : 41–56. doi : 10.1093/bmb/ldt021 . PMID 23896486.
- ^ abc Merlini G, Dispenzieri A, Sanchorawala V, Schönland SO, Palladini G, Hawkins PN, Gertz MA (octubre de 2018). "Amiloidosis sistémica de cadenas ligeras de inmunoglobulinas". Reseñas de la naturaleza. Cebadores de enfermedades . 4 (1): 38. doi :10.1038/s41572-018-0034-3. PMID 30361521. S2CID 53023121. Archivado desde el original el 14 de junio de 2022 . Consultado el 25 de diciembre de 2020 .
- ^ ab Wechalekar AD, Gillmore JD, Hawkins PN (junio de 2016). "Amiloidosis sistémica". Lancet . 387 (10038): 2641–2654. doi :10.1016/S0140-6736(15)01274-X. PMID 26719234. S2CID 4762107.
- ^ ab Rosenzweig M, Landau H (noviembre de 2011). "Amiloidosis de cadena ligera (AL): actualización sobre diagnóstico y tratamiento". Journal of Hematology & Oncology . 4 (1): 47. doi : 10.1186/1756-8722-4-47 . PMC 3228694 . PMID 22100031.
- ^ Mok KH, Pettersson J, Orrenius S, Svanborg C (marzo de 2007). "HAMLET, plegamiento de proteínas y muerte de células tumorales". Comunicaciones de investigación bioquímica y biofísica . 354 (1): 1–7. doi :10.1016/j.bbrc.2006.12.167. PMID 17223074.
- ^ Pettersson-Kastberg J, Aits S, Gustafsson L, Mossberg A, Storm P, Trulsson M, et al. (noviembre de 2008). "¿Pueden ser beneficiosas las proteínas mal plegadas? El caso HAMLET". Annals of Medicine . 41 (3): 162–176. doi :10.1080/07853890802502614. PMID 18985467. S2CID 31198109.
- ^ Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJ, Sekijima Y, et al. (diciembre de 2020). "Nomenclatura de amiloide 2020: actualización y recomendaciones del comité de nomenclatura de la Sociedad Internacional de Amiloidosis (ISA)". Amiloide . 27 (4): 217–222. doi : 10.1080/13506129.2020.1835263 . PMID 33100054. S2CID 225073269.
- ^ abc Tabla 5-12 en: Mitchell RS, Kumar V, Abbas AK, Fausto N (2007). Robbins Basic Pathology . Filadelfia: Saunders. ISBN 978-1-4160-2973-1.8va edición.
- ^ ter Haar NM, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA, et al. (septiembre de 2015). "Recomendaciones para el tratamiento de enfermedades autoinflamatorias". Anales de las enfermedades reumáticas . 74 (9): 1636–1644. doi :10.1136/annrheumdis-2015-207546. hdl : 2445/108897 . PMID 26109736. S2CID 18876892.
- ^ abc Magrinelli F, Fabrizi GM, Santoro L, Manganelli F, Zanette G, Cavallaro T, Tamburin S, et al. (Grupo Cochrane Neuromuscular) (abril de 2020). "Tratamiento farmacológico para la polineuropatía amiloide familiar". Base de datos Cochrane de revisiones sistemáticas . 4 (4): CD012395. doi :10.1002/14651858.CD012395.pub2. PMC 7170468. PMID 32311072 .
- ^ ab Marques N, Azevedo O, Almeida AR, Bento D, Cruz I, Correia E, et al. (octubre de 2020). "Terapia específica para la amiloidosis cardíaca por transtiretina: una revisión sistemática de la literatura y recomendaciones basadas en la evidencia". Revista de la Asociación Estadounidense del Corazón . 9 (19): e016614. doi :10.1161/JAHA.120.016614. PMC 7792401 . PMID 32969287.
- ^ "Tegsedi". Agencia Europea de Medicamentos . Archivado desde el original el 8 de octubre de 2020. Consultado el 12 de marzo de 2021 .
- ^ Mathew V, Wang AK (6 de mayo de 2019). "Inotersen: una nueva promesa para el tratamiento de la amiloidosis hereditaria por transtiretina". Diseño, desarrollo y terapia de fármacos . 13 : 1515–1525. doi : 10.2147/DDDT.S162913 . PMC 6507904. PMID 31118583 .
- ^ "Patisiran para el tratamiento de la amiloidosis hereditaria relacionada con la transtiretina". Archivado desde el original el 4 de julio de 2019. Consultado el 20 de julio de 2019 .
- ^ "Patisiran para el tratamiento de la amiloidosis hereditaria relacionada con la transtiretina [ID1279] | Guía | NICE". Instituto Nacional para la Excelencia en la Salud y la Atención (Nice) . Archivado desde el original el 20 de julio de 2019. Consultado el 20 de julio de 2019 .
- ^ Hoy SM (octubre de 2018). "Patisiran: primera aprobación global". Drugs . 78 (15): 1625–1631. doi :10.1007/s40265-018-0983-6. PMID 30251172. S2CID 52813638.
- ^ Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, et al. (agosto de 2021). "Edición genética in vivo con CRISPR-Cas9 para la amiloidosis por transtiretina". The New England Journal of Medicine . 385 (6): 493–502. doi : 10.1056/NEJMoa2107454 . PMID 34215024. S2CID 235722446.
- ^ "Alnylam anuncia la aprobación por parte de la FDA de Amvuttra (vutrisiran), un fármaco terapéutico de ARNi para el tratamiento de la polineuropatía de la amiloidosis hereditaria mediada por transtiretina en adultos". Alnylam. 13 de junio de 2022. Archivado desde el original el 14 de junio de 2022. Consultado el 14 de junio de 2022 a través de Business Wire.
- ^ "Amiloidosis - NORD (Organización Nacional de Enfermedades Raras)". NORD (Organización Nacional de Enfermedades Raras) . Archivado desde el original el 16 de marzo de 2016. Consultado el 15 de marzo de 2016 .
- ^ "Amiloidosis cutánea primaria – Enfermedad – Organizaciones – Centro de información sobre enfermedades genéticas y raras (GARD) – Programa NCATS". rarediseases.info.nih.gov . Archivado desde el original el 15 de marzo de 2016 . Consultado el 15 de marzo de 2016 .
- ^ Merlini G (diciembre de 2017). "Amiloidosis AL: de mecanismos moleculares a terapias dirigidas". Hematología. Sociedad Estadounidense de Hematología. Programa Educativo . 2017 (1): 1–12. doi :10.1182/asheducation-2017.1.1. PMC 6142527. PMID 29222231 .
- ^ ab Falk RH, Alexander KM, Liao R, Dorbala S (septiembre de 2016). "Amiloidosis cardíaca de cadena ligera (AL): una revisión del diagnóstico y la terapia". Revista del Colegio Americano de Cardiología . 68 (12): 1323–1341. doi : 10.1016/j.jacc.2016.06.053 . PMID 27634125.
- ^ ab Xin Y, Hu W, Chen X, Hu J, Sun Y, Zhao Y (noviembre de 2019). "Impacto pronóstico de las categorías relacionadas con la cadena ligera y la transtiretina en la amiloidosis cardíaca: una revisión sistemática y un metanálisis". Revista helénica de cardiología . 60 (6): 375–383. doi : 10.1016/j.hjc.2019.01.015 . PMID 30742933. S2CID 73419672.
- ^ Coles LS, Young RD (mayo de 2012). "Supercentenarios y amiloidosis por transtiretina: la próxima frontera de la prolongación de la vida humana". Medicina preventiva . 54 (Supl.): S9-11. doi :10.1016/j.ypmed.2012.03.003. PMID 22579241.
- ^ "Buscando los secretos de los superviejos". Science. 26 de septiembre de 2008. pp. 1764–1765. Archivado desde el original el 9 de marzo de 2013. Consultado el 22 de febrero de 2013 .
- ^ ab Lin HM, Gao X, Cooke CE, Berg D, Labotka R, Faller DV, et al. (junio de 2017). "Carga de enfermedad de la amiloidosis sistémica de cadena ligera: una revisión sistemática de la literatura". Investigación médica actual y opinión . 33 (6): 1017–1031. doi :10.1080/03007995.2017.1297930. PMID 28277869. S2CID 205541963.
- ^ Brunger AF, Nienhuis HL, Bijzet J, Hazenberg BP (marzo de 2020). "Causas de la amiloidosis AA: una revisión sistemática". Amiloide . 27 (1): 1–12. doi : 10.1080/13506129.2019.1693359 . PMID 31766892. S2CID 208299494.
- ^ Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, Singleton A, Kiuru-Enari S, Paetau A, Tienari PJ, Myllykangas L (1 de enero de 2008). "La amiloidosis sistémica senil afecta al 25% de las personas muy mayores y se asocia con variación genética en alfa2-macroglobulina y tau: un estudio de autopsia basado en la población". Annals of Medicine . 40 (3): 232–239. doi :10.1080/07853890701842988. ISSN 0785-3890. PMID 18382889. S2CID 23446885. Archivado desde el original el 14 de junio de 2022 . Consultado el 18 de marzo de 2022 .
- ^ Hasib Sidiqi M, Gertz MA (mayo de 2021). "Algoritmo de diagnóstico y tratamiento de la amiloidosis de cadena ligera de inmunoglobulina 2021". Blood Cancer Journal . 11 (5): 90. doi :10.1038/s41408-021-00483-7. PMC 8124067 . PMID 33993188.
- ^ Buxbaum JN (julio de 2018). "Fármacos oligonucleótidos para la amiloidosis por transtiretina". The New England Journal of Medicine . 379 (1): 82–85. doi :10.1056/nejme1805499. PMID 29972750. S2CID 49658028.
- ^ Oficina del Comisionado. "Comunicados de prensa: la FDA aprueba la primera terapia dirigida basada en ARN de su tipo para tratar una enfermedad rara". www.fda.gov . Archivado desde el original el 7 de septiembre de 2018 . Consultado el 11 de agosto de 2018 .
- ^ ab Cristóbal Gutiérrez H, Pelayo-Negro AL, Gómez Gómez D, Martín Vega MÁ, Valero Domínguez M (julio de 2020). "Resumen de los tratamientos utilizados en la amiloidosis hereditaria relacionada con la transtiretina: una revisión sistemática". Revista Europea de Farmacia Hospitalaria . 27 (4): 194-201. doi :10.1136/ejhpharm-2018-001823. PMC 7335620 . PMID 32587078.
Enlaces externos
