Sistema simplificado de entrada de línea de entrada molecular
| Extensión de nombre de archivo | .smi |
|---|---|
| Tipo de medio de Internet | química/x-luz-del-día-sonrisas |
| Tipo de formato | formato de archivo químico |

El sistema de entrada de línea de entrada molecular simplificado ( SMILES ) es una especificación en forma de notación de línea para describir la estructura de especies químicas utilizando cadenas ASCII cortas. La mayoría de los editores de moléculas pueden importar cadenas SMILES para convertirlas nuevamente en dibujos bidimensionales o modelos tridimensionales de las moléculas.
La especificación SMILES original se inició en la década de 1980. Desde entonces, se ha modificado y ampliado. En 2007, se desarrolló un estándar abierto llamado OpenSMILES en la comunidad de química de código abierto .
Historia
La especificación original de SMILES fue iniciada por David Weininger en el Laboratorio de la División de Ecología del Continente Medio de la USEPA en Duluth en la década de 1980. [1] [2] [3] [4] Se reconoció por su participación en el desarrollo inicial a "Gilman Veith y Rose Russo (USEPA) y Albert Leo y Corwin Hansch ( Pomona College ) por apoyar el trabajo, y a Arthur Weininger (Pomona; Daylight CIS) y Jeremy Scofield (Cedar River Software, Renton, WA) por su ayuda en la programación del sistema". [5] La Agencia de Protección Ambiental financió el proyecto inicial para desarrollar SMILES. [6] [7]
Desde entonces, otros lo han modificado y ampliado, en particular Daylight Chemical Information Systems. En 2007, la comunidad de química de código abierto Blue Obelisk desarrolló un estándar abierto llamado "OpenSMILES". Otras notaciones "lineales" incluyen Wiswesser Line Notation (WLN), ROSDAL y SLN (Tripos Inc).
En julio de 2006, la IUPAC introdujo el InChI como estándar para la representación de fórmulas. En general, se considera que SMILES tiene la ventaja de ser más legible para los humanos que el InChI; también cuenta con una amplia base de soporte de software con un amplio respaldo teórico (como la teoría de grafos ).
Terminología
El término SMILES se refiere a una notación lineal para codificar estructuras moleculares y las instancias específicas deben llamarse estrictamente cadenas SMILES. Sin embargo, el término SMILES también se usa comúnmente para referirse tanto a una sola cadena SMILES como a varias cadenas SMILES; el significado exacto suele ser evidente a partir del contexto. Los términos "canónico" e "isomérico" pueden generar cierta confusión cuando se aplican a SMILES. Los términos describen diferentes atributos de las cadenas SMILES y no son mutuamente excluyentes.
Por lo general, se pueden escribir varias cadenas SMILES igualmente válidas para una molécula. Por ejemplo, CCO, OCCy C(O)Ctodas especifican la estructura del etanol . Se han desarrollado algoritmos para generar la misma cadena SMILES para una molécula dada; de las muchas cadenas posibles, estos algoritmos eligen solo una de ellas. Este SMILES es único para cada estructura, aunque depende del algoritmo de canonización utilizado para generarlo, y se denomina SMILES canónico. Estos algoritmos primero convierten el SMILES en una representación interna de la estructura molecular; luego, un algoritmo examina esa estructura y produce una cadena SMILES única. Se han desarrollado varios algoritmos para generar SMILES canónicos, entre ellos los de Daylight Chemical Information Systems, OpenEye Scientific Software , MEDIT, Chemical Computing Group , MolSoft LLC y Chemistry Development Kit . Una aplicación común de SMILES canónico es indexar y garantizar la unicidad de las moléculas en una base de datos .
El artículo original que describió el algoritmo CANGEN [2] afirmó generar cadenas SMILES únicas para gráficos que representan moléculas, pero el algoritmo falla en varios casos simples (por ejemplo, cuneano , 1,2-diciclopropiletano) y no puede considerarse un método correcto para representar un gráfico canónicamente. [8] Actualmente no existe una comparación sistemática entre software comercial para probar si existen tales fallas en esos paquetes.
La notación SMILES permite especificar la configuración en los centros tetraédricos y la geometría del doble enlace. Estas son características estructurales que no se pueden especificar solo mediante la conectividad y, por lo tanto, los SMILES que codifican esta información se denominan SMILES isoméricos. Una característica notable de estas reglas es que permiten una especificación parcial rigurosa de la quiralidad. El término SMILES isomérico también se aplica a los SMILES en los que se especifican los isómeros .
Definición basada en gráficos
En términos de un procedimiento computacional basado en gráficos, SMILES es una cadena que se obtiene al imprimir los nodos de símbolos encontrados en un recorrido de árbol en profundidad de un gráfico químico . El gráfico químico primero se recorta para eliminar átomos de hidrógeno y se rompen los ciclos para convertirlo en un árbol de expansión . Cuando se han roto los ciclos, se incluyen etiquetas de sufijo numérico para indicar los nodos conectados. Los paréntesis se utilizan para indicar puntos de ramificación en el árbol.
La forma SMILES resultante depende de las opciones elegidas:
- de los vínculos elegidos para romper ciclos,
- del átomo de partida utilizado para el recorrido en profundidad, y
- del orden en que se enumeran las ramas cuando se encuentran.
Definición de SMILES como cadenas de un lenguaje libre de contexto
Desde el punto de vista de una teoría del lenguaje formal, SMILES es una palabra. Un SMILES se puede analizar con un analizador sintáctico independiente del contexto. El uso de esta representación se ha realizado en la predicción de propiedades bioquímicas (incluida la toxicidad y la biodegradabilidad ) basándose en el principio fundamental de la quimioinformática de que las moléculas similares tienen propiedades similares. Los modelos predictivos implementaron un enfoque de reconocimiento de patrones sintácticos (que implicaba definir una distancia molecular) [9] , así como un esquema más robusto basado en el reconocimiento de patrones estadísticos. [10]
Descripción
Átomos
Los átomos se representan mediante la abreviatura estándar de los elementos químicos , entre corchetes, como en [Au]el caso del oro . Los corchetes pueden omitirse en el caso común de átomos que:
- están en el " subconjunto orgánico " de B , C , N , O , P , S , F , Cl , Br o I , y
- no tienen cargo formal , y
- tienen el número de hidrógenos unidos implícito en el modelo de valencia SMILES (normalmente su valencia normal, pero para N y P es 3 o 5, y para S es 2, 4 o 6), y
- son los isótopos normales , y
- no son centros quirales .
Todos los demás elementos deben estar entre corchetes y sus cargas e hidrógenos deben indicarse explícitamente. Por ejemplo, el SMILES para el agua puede escribirse como Oo [OH2]. El hidrógeno también puede escribirse como un átomo separado; el agua también puede escribirse como [H]O[H].
Cuando se utilizan corchetes, Hse añade el símbolo si el átomo entre corchetes está unido a uno o más hidrógenos, seguido del número de átomos de hidrógeno si es mayor que 1, luego del signo +para una carga positiva o -para una carga negativa. Por ejemplo, [NH4+]para el amonio ( NH+
4). Si hay más de una carga, normalmente se escribe como dígito; sin embargo, también es posible repetir el signo tantas veces como cargas tenga el ion: se puede escribir [Ti+4]o [Ti++++]para el titanio (IV) Ti 4+ . Así, el anión hidróxido ( OH − ) se representa por , el catión hidronio ( H 3 O + ) es y el catión cobalto (III) (Co 3+ ) es o . [OH-][OH3+][Co+3][Co+++]
Cautiverio
Un enlace se representa mediante uno de los símbolos . - = # $ : / \.
Se supone que los enlaces entre átomos alifáticos son simples a menos que se especifique lo contrario y están implícitos por la adyacencia en la cadena SMILES. Aunque los enlaces simples pueden escribirse como -, esto se omite generalmente. Por ejemplo, el SMILES para etanol puede escribirse como C-C-O, CC-Oo C-CO, pero generalmente se escribe CCO.
Los enlaces dobles, triples y cuádruples se representan con los símbolos =, #, y $respectivamente, como lo ilustra SMILES O=C=O( dióxido de carbono CO 2 ), C#N( cianuro de hidrógeno HCN) y [Ga+]$[As-]( arseniuro de galio ).
Un tipo adicional de enlace es un "no enlace", indicado con ., para indicar que dos partes no están unidas entre sí. Por ejemplo, el cloruro de sodio acuoso puede escribirse como [Na+].[Cl-]para mostrar la disociación.
Un enlace aromático "uno y medio" puede indicarse con :; ver § Aromaticidad a continuación.
Los enlaces simples adyacentes a enlaces dobles pueden representarse utilizando /o \para indicar la configuración estereoquímica; consulte § Estereoquímica a continuación.
Anillos
Las estructuras de anillo se escriben rompiendo cada anillo en un punto arbitrario (aunque algunas opciones darán lugar a un SMILES más legible que otras) para crear una estructura acíclica y agregar etiquetas numéricas de cierre de anillo para mostrar la conectividad entre átomos no adyacentes.
Por ejemplo, el ciclohexano y el dioxano pueden escribirse como C1CCCCC1y O1CCOCC1respectivamente. Para un segundo anillo, la etiqueta será 2. Por ejemplo, la decalina (decahidronaftaleno) puede escribirse como C1CCCC2C1CCCC2.
SMILES no requiere que los números de anillo se utilicen en un orden particular y permite el número de anillo cero, aunque esto rara vez se usa. Además, se permite reutilizar los números de anillo después de que se haya cerrado el primer anillo, aunque esto generalmente hace que las fórmulas sean más difíciles de leer. Por ejemplo, biciclohexilo generalmente se escribe como C1CCCCC1C2CCCCC2, pero también puede escribirse como C0CCCCC0C0CCCCC0.
Varios dígitos después de un solo átomo indican múltiples enlaces de cierre de anillo. Por ejemplo, una notación SMILES alternativa para la decalina es C1CCCC2CCCCC12, donde el carbono final participa en ambos enlaces de cierre de anillo 1 y 2. Si se requieren números de anillo de dos dígitos, la etiqueta está precedida por %, por lo que C%12se trata de un solo enlace de cierre de anillo del anillo 12.
Cualquiera o ambos dígitos pueden estar precedidos por un tipo de enlace para indicar el tipo de enlace de cierre de anillo. Por ejemplo, el ciclopropeno suele escribirse como C1=CC1, pero si se elige el doble enlace como enlace de cierre de anillo, puede escribirse como C=1CC1, C1CC=1, o C=1CC=1. (Se prefiere la primera forma). C=1CC-1es ilegal, ya que especifica explícitamente tipos conflictivos para el enlace de cierre de anillo.
Los enlaces de cierre de anillo no se pueden utilizar para denotar enlaces múltiples. Por ejemplo, C1C1no es una alternativa válida para C=Cel etileno . Sin embargo, se pueden utilizar con enlaces no convencionales; C1.C2.C12es una forma alternativa peculiar pero legal de escribir propano , que se escribe más comúnmente como CCC.
La elección de un punto de ruptura del anillo adyacente a los grupos unidos puede dar lugar a una forma SMILES más simple al evitar las ramificaciones. Por ejemplo, el ciclohexano-1,2-diol se escribe de forma más sencilla como OC1CCCCC1O; la elección de una ubicación de ruptura del anillo diferente produce una estructura ramificada que requiere paréntesis para escribirse.
Aromaticidad
Los anillos aromáticos como el benceno pueden escribirse en una de tres formas:
- En forma de Kekulé con enlaces simples y dobles alternados, por ejemplo
C1=CC=CC=C1, - Utilizando el símbolo de enlace aromático
:, por ejemploC:1:C:C:C:C:C1, o - Lo más común es escribir los átomos constituyentes B, C, N, O, P y S en minúsculas
b,c,n, y , respectivamente.ops
En el último caso, se supone que los enlaces entre dos átomos aromáticos (si no se muestra explícitamente) son enlaces aromáticos. Por lo tanto, el benceno , la piridina y el furano pueden representarse respectivamente mediante los símbolos SMILES c1ccccc1y .n1ccccc1o1cccc1
El nitrógeno aromático unido al hidrógeno, como el que se encuentra en el pirrol, debe representarse como [nH]; por lo tanto, el imidazol se escribe en notación SMILES como n1c[nH]cc1.
Cuando los átomos aromáticos están unidos entre sí mediante enlaces simples, como en el caso del bifenilo , se debe mostrar explícitamente un enlace simple: c1ccccc1-c2ccccc2. Este es uno de los pocos casos en los que -se requiere el símbolo de enlace simple. (De hecho, la mayoría del software SMILES puede inferir correctamente que el enlace entre los dos anillos no puede ser aromático y, por lo tanto, aceptará la forma no estándar c1ccccc1c2ccccc2).
Los algoritmos Daylight y OpenEye para generar SMILES canónicos difieren en su tratamiento de la aromaticidad.

COc(c1)cccc1C#N.Derivación
Las ramificaciones se describen con paréntesis, como en el caso CCC(=O)Odel ácido propiónico y FC(F)Fdel fluoroformo . El primer átomo dentro de los paréntesis y el primer átomo después del grupo entre paréntesis están unidos al mismo átomo del punto de ramificación. El símbolo de enlace debe aparecer dentro de los paréntesis; fuera de ellos (p. ej.: CCC=(O)O) no es válido.
Los anillos sustituidos se pueden escribir con el punto de ramificación en el anillo, como se ilustra en los SMILES COc(c1)cccc1C#N(ver la ilustración) y COc(cc1)ccc1C#N(ver la ilustración), que codifican los isómeros 3 y 4-cianoanisol. Escribir SMILES para anillos sustituidos de esta manera puede hacerlos más legibles para los humanos.
Las ramas se pueden escribir en cualquier orden. Por ejemplo, bromoclorodifluorometano se puede escribir como FC(Br)(Cl)F, BrC(F)(F)Cl, C(F)(Cl)(F)Br, o algo similar. En general, una forma SMILES es más fácil de leer si la rama más simple va primero, y la parte final, sin paréntesis, es la más compleja. Las únicas advertencias para tales reordenamientos son:
- Si se reutilizan los números de anillo, se emparejan según el orden en que aparecen en la cadena SMILES. Es posible que se requieran algunos ajustes para preservar el emparejamiento correcto.
- Si se especifica estereoquímica, se deben realizar ajustes; consulte § Estereoquímica a continuación.
La única forma de ramificación que no requiere paréntesis son los enlaces de cierre de anillo: el fragmento SMILES C1Nes equivalente a C(1)N, ambos denotan un enlace entre el Cy el N. La elección de enlaces de cierre de anillo adyacentes a los puntos de ramificación puede reducir la cantidad de paréntesis necesarios. Por ejemplo, el tolueno normalmente se escribe como Cc1ccccc1o c1ccccc1C, evitando los paréntesis necesarios si se escribe como c1cc(C)ccc1o c1cc(ccc1)C.
Estereoquímica
SMILES permite, pero no requiere, la especificación de estereoisómeros .
La configuración en torno a los enlaces dobles se especifica utilizando los caracteres /y \para mostrar enlaces simples direccionales adyacentes a un enlace doble. Por ejemplo, F/C=C/F(ver la representación) es una representación de trans - 1,2-difluoroetileno , en la que los átomos de flúor están en lados opuestos del doble enlace (como se muestra en la figura), mientras que F/C=C\F(ver la representación) es una posible representación de cis -1,2-difluoroetileno, en la que los átomos de flúor están en el mismo lado del doble enlace.
Los símbolos de dirección de enlace siempre vienen en grupos de al menos dos, de los cuales el primero es arbitrario. Es decir, F\C=C\Fes igual a F/C=C/F. Cuando hay enlaces simples y dobles alternados, los grupos son mayores que dos, y los símbolos direccionales del medio están adyacentes a dos enlaces dobles. Por ejemplo, la forma común de (2,4)-hexadieno se escribe C/C=C/C=C/C.

Como ejemplo más complejo, el betacaroteno tiene una cadena principal muy larga de enlaces simples y dobles alternados, que puede escribirse como CC1CCC/C(C)=C1/C=C/C(C)=C/C=C/C(C)=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C2=C(C)/CCCC2(C)C.
La configuración en el carbono tetraédrico se especifica con @o @@. Considere los cuatro enlaces en el orden en que aparecen, de izquierda a derecha, en la forma SMILES. Mirando hacia el carbono central desde la perspectiva del primer enlace, los otros tres están en el sentido de las agujas del reloj o en el sentido contrario a las agujas del reloj. Estos casos se indican con @@y @, respectivamente (porque el @símbolo en sí es una espiral en sentido contrario a las agujas del reloj).

Por ejemplo, considere el aminoácido alanina . Una de sus formas SMILES es NC(C)C(=O)O, escrita de forma más completa como N[CH](C)C(=O)O. L -alanina , el enantiómero más común , se escribe como N[C@@H](C)C(=O)O(ver la ilustración). Mirando desde el enlace nitrógeno-carbono, los grupos hidrógeno ( H), metilo ( C) y carboxilato ( C(=O)O) aparecen en el sentido de las agujas del reloj. La D -alanina se puede escribir como N[C@H](C)C(=O)O(ver la ilustración).
Si bien el orden en el que se especifican las ramas en SMILES normalmente no es importante, en este caso sí lo es; intercambiar dos grupos cualesquiera requiere invertir el indicador de quiralidad. Si las ramas se invierten de modo que la alanina se escriba como NC(C(=O)O)C, entonces la configuración también se invierte; la L -alanina se escribe como N[C@H](C(=O)O)C(ver la representación). Otras formas de escribirlo incluyen C[C@H](N)C(=O)O, OC(=O)[C@@H](N)Cy OC(=O)[C@H](C)N.
Normalmente, el primero de los cuatro enlaces aparece a la izquierda del átomo de carbono, pero si el SMILES se escribe empezando por el carbono quiral, como por ejemplo C(C)(N)C(=O)O, entonces los cuatro están a la derecha, pero el primero que aparece (el [CH]enlace en este caso) se utiliza como referencia para ordenar los tres siguientes: L -alanina también puede escribirse [C@@H](C)(N)C(=O)O.
La especificación SMILES incluye elaboraciones sobre el @símbolo para indicar la estereoquímica alrededor de centros quirales más complejos, como la geometría molecular bipiramidal trigonal .
Isótopos
Los isótopos se especifican con un número igual a la masa isotópica entera que precede al símbolo atómico. El benceno , en el que un átomo es carbono-14, se escribe como [14c]1ccccc1y el deuterocloroformo como [2H]C(Cl)(Cl)Cl.
Ejemplos
| Molécula | Estructura | Fórmula SONRISAS |
|---|---|---|
| Dinitrógeno | N≡N | N#N |
| Isocianato de metilo (MIC) | CH3 - N =C=O | CN=C=O |
| Sulfato de cobre (II) | Cu2 + SO42− 4 | [Cu+2].[O-]S(=O)(=O)[O-] |
| Vanilina |  | O=Cc1ccc(O)c(OC)c1COc1cc(C=O)ccc1O |
| Melatonina (C 13 H 16 N 2 O 2 ) |  | CC(=O)NCCC1=CNc2c1cc(OC)cc2CC(=O)NCCc1c[nH]c2ccc(OC)cc12 |
| Flavopereirina (C 17 H 15 N 2 ) |  | CCc(c1)ccc2[n+]1ccc3c2[nH]c4c3cccc4CCc1c[n+]2ccc3c4ccccc4[nH]c3c2cc1 |
| Nicotina (C 10 H 14 N 2 ) |  | CN1CCC[C@H]1c2cccnc2 |
| Enantotoxina (C 17 H 22 O 2 ) |  | CCC[C@@H](O)CC\C=C\C=C\C#CC#C\C=C\COCCC[C@@H](O)CC/C=C/C=C/C#CC#C/C=C/CO |
| Piretrina II (C 22 H 28 O 5 ) |  | CC1=C(C(=O)C[C@@H]1OC(=O)[C@@H]2[C@H](C2(C)C)/C=C(\C)/C(=O)OC)C/C=C\C=C |
| Aflatoxina B1 ( C17H12O6 ) | 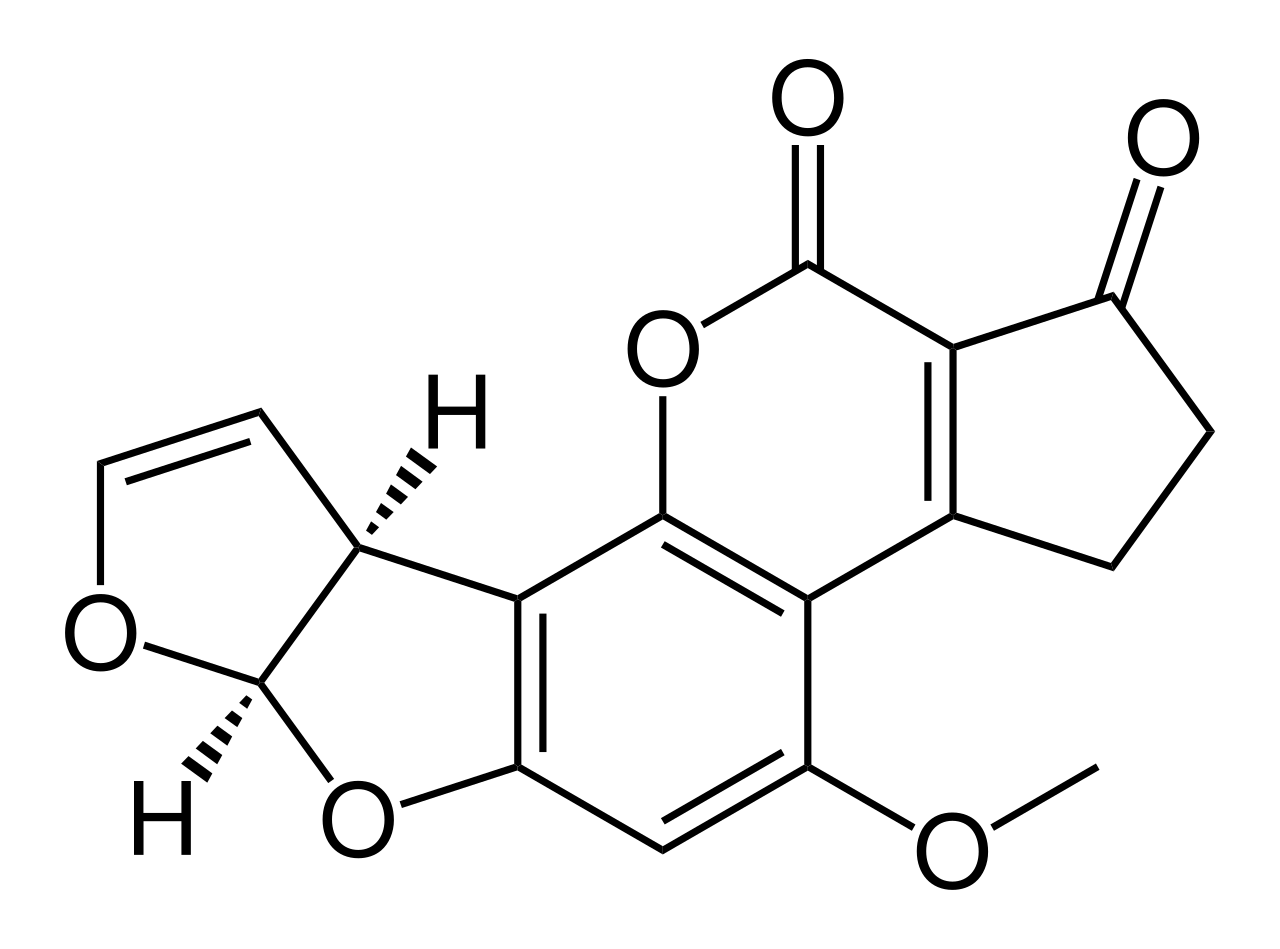 | O1C=C[C@H]([C@H]1O2)c3c2cc(OC)c4c3OC(=O)C5=C4CCC(=O)5 |
| Glucosa (β- D -glucopiranosa) (C 6 H 12 O 6 ) | 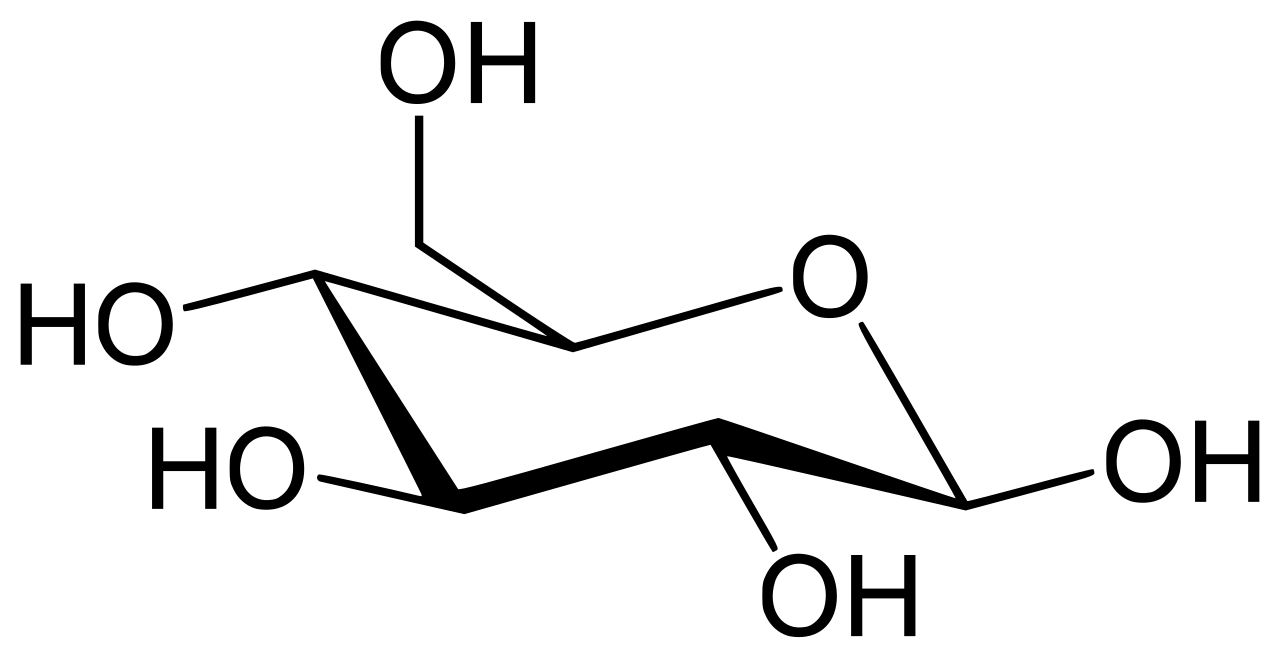 | OC[C@@H](O1)[C@@H](O)[C@H](O)[C@@H](O)[C@H](O)1 |
| Bergenina (cuscutina, una resina ) (C 14 H 16 O 9 ) |  | OC[C@@H](O1)[C@@H](O)[C@H](O)[C@@H]2[C@@H]1c3c(O)c(OC)c(O)cc3C(=O)O2 |
| Una feromona de la cochinilla californiana |  | CC(=O)OCCC(/C)=C\C[C@H](C(C)=C)CCC=C |
| (2 S ,5 R )-Chalcogran: una feromona del gorgojo de la corteza Pityogenes chalcographus [11] | ![(2S,5R)-2-etil-1,6-dioxaspiro[4.4]nonano](http://upload.wikimedia.org/wikipedia/commons/thumb/8/8e/2S,5R-chalcogran-skeletal.svg/1280px-2S,5R-chalcogran-skeletal.svg.png) | CC[C@H](O1)CC[C@@]12CCCO2 |
| α-Tuyona (C 10 H 16 O) |  | CC(C)[C@@]12C[C@@H]1[C@@H](C)C(=O)C2 |
| Tiamina ( vitamina B1 , C12H17N4OS + ) |  | OCCc1c(C)[n+](cs1)Cc2cnc(C)nc2N |
Para ilustrar una molécula con más de 9 anillos, considere la cefalostatina-1, [12] una pirazina esteroidal de 13 anillos con la fórmula empírica C 54 H 74 N 2 O 10 aislada del hemicordado del Océano Índico Cephalodiscus gilchristi :

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comenzando con el grupo metilo más a la izquierda en la figura:
CC(C)(O1)C[C@@H](O)[C@@]1(O2)[C@@H](C)[C@@H]3CC=C4[C@]3(C2)C(=O)C[C@H]5[C@H]4CC[C@@H](C6)[C@]5(C)Cc(n7)c6nc(C[C@@]89(C))c7C[C@@H]8CC[C@@H]%10[C@@H]9C[C@@H](O)[C@@]%11(C)C%10=C[C@H](O%12)[C@]%11(O)[C@H](C)[C@]%12(O%13)[C@H](O)C[C@@]%13(C)CO
%aparece delante del índice de etiquetas de cierre de anillos arriba 9; ver § Anillos arriba.
Otros ejemplos de SONRISAS
La notación SMILES se describe en detalle en el manual de teoría SMILES proporcionado por Daylight Chemical Information Systems y se presentan varios ejemplos ilustrativos. La utilidad de representación de Daylight ofrece a los usuarios los medios para verificar sus propios ejemplos de SMILES y es una valiosa herramienta educativa.
Extensiones
SMARTS es una notación lineal para la especificación de patrones subestructurales en moléculas. Si bien utiliza muchos de los mismos símbolos que SMILES, también permite la especificación de átomos y enlaces comodín , que se pueden utilizar para definir consultas subestructurales para la búsqueda en bases de datos químicas . Un error común es que la búsqueda subestructural basada en SMARTS implica la coincidencia de cadenas SMILES y SMARTS. De hecho, tanto las cadenas SMILES como SMARTS se convierten primero en representaciones gráficas internas en las que se busca el isomorfismo de subgrafos .
SMIRKS, un superconjunto de "reaction SMILES" y un subconjunto de "reaction SMARTS", es una notación de línea para especificar transformaciones de reacción. La sintaxis general para las extensiones de reacción es REACTANT>AGENT>PRODUCT(sin espacios), donde cualquiera de los campos puede dejarse en blanco o rellenarse con múltiples moléculas delineadas con un punto ( .), y otras descripciones que dependen del lenguaje base. Los átomos pueden identificarse adicionalmente con un número (por ejemplo [C:1]) para su mapeo, [13] por ejemplo en . [14]
SMILES corresponde a estructuras moleculares discretas. Sin embargo, muchos materiales son macromoléculas, que son demasiado grandes (y a menudo estocásticas) para generar SMILES de manera conveniente. BigSMILES es una extensión de SMILES que tiene como objetivo proporcionar un sistema de representación eficiente para macromoléculas. [15]
Conversión
Los SMILES se pueden convertir nuevamente a representaciones bidimensionales mediante algoritmos de generación de diagramas de estructura (SDG). [16] Esta conversión a veces es ambigua. La conversión a una representación tridimensional se logra mediante enfoques de minimización de energía. Existen muchas utilidades de conversión descargables y basadas en la web.
Véase también
- Especificación de objetivo arbitrario SMILES (SMARTS), una extensión de SMILES para la especificación de consultas subestructurales
- Notación de línea SYBYL , otra notación de línea
- Identificador químico internacional (InChI), la alternativa de la IUPAC a SMILES
- Lenguaje de consulta molecular , un lenguaje de consulta que también permite propiedades numéricas, por ejemplo, valores fisicoquímicos o distancias.
- Kit de desarrollo de química , software de diseño y conversión 2D
- OpenBabel , JOELib , OELib (conversión)
Referencias
- ^ Weininger D (febrero de 1988). "SMILES, un lenguaje químico y un sistema de información. 1. Introducción a la metodología y a las reglas de codificación". Revista de información química y ciencias de la computación . 28 (1): 31–6. doi :10.1021/ci00057a005.
- ^ ab Weininger D, Weininger A, Weininger JL (mayo de 1989). "SMILES. 2. Algoritmo para la generación de notación SMILES única". Revista de información y modelado químico . 29 (2): 97–101. doi :10.1021/ci00062a008.
- ^ Weininger D (agosto de 1990). "SMILES. 3. DEPICT. Representación gráfica de estructuras químicas". Revista de información y modelado químico . 30 (3): 237–43. doi :10.1021/ci00067a005.
- ^ Swanson RP (2004). "La entrada de la informática en la química combinatoria" (PDF) . En Rayward WB, Bowden ME (eds.). La historia y el legado de los sistemas de información científica y tecnológica: Actas de la Conferencia de 2002 de la Sociedad Estadounidense de Ciencias de la Información y Tecnología y la Fundación del Patrimonio Químico. Medford, NJ: Information Today . p. 205. ISBN. 978-1-57387-229-4.
- ^ Weininger D (1998). "Agradecimientos en la página de sonrisas, etc. del Tutorial de luz diurna" . Consultado el 24 de junio de 2013 .
- ^ Anderson E, Veith GD, Weininger D (1987). SMILES: una notación lineal e intérprete computarizado para estructuras químicas (PDF) . Duluth, MN: US EPA , Laboratorio de Investigación Ambiental-Duluth. Informe n.º EPA/600/M-87/021.
- ^ "Tutorial de SMILES: ¿Qué es SMILES?". US EPA . Archivado desde el original el 28 de marzo de 2008. Consultado el 23 de septiembre de 2012 .
- ^ Neglur G, Grossman RL, Liu B (2005). "Asignación de claves únicas a compuestos químicos para la integración de datos: algunos ejemplos interesantes". En Ludäscher B (ed.). Integración de datos en las ciencias de la vida . Apuntes de clase en informática. Vol. 3615. Berlín: Springer. págs. 145–157. doi :10.1007/11530084_13. ISBN. 978-3-540-27967-9. Recuperado el 12 de febrero de 2013 .
- ^ Sidorova J, Anisimova M (agosto de 2014). "Reconocimiento de patrones estructurales inspirado en PNL en aplicaciones químicas". Pattern Recognition Letters . 45 : 11–16. Código Bibliográfico :2014PaReL..45...11S. doi :10.1016/j.patrec.2014.02.012.
- ^ Sidorova J, Garcia J (noviembre de 2015). "Un puente entre los métodos sintácticos y los estadísticos: clasificación con características segmentadas automáticamente a partir de secuencias". Reconocimiento de patrones . 48 (11): 3749–3756. Bibcode :2015PatRe..48.3749S. doi :10.1016/j.patcog.2015.05.001. hdl : 10016/33552 .
- ^ Byers JA, Birgersson G, Löfqvist J, Appelgren M, Bergström G (marzo de 1990). "Aislamiento de sinergistas de feromonas del gorgojo de la corteza, Pityogenes chalcographus, a partir de olores complejos de insectos y plantas mediante fraccionamiento y bioensayo de combinación sustractiva". Journal of Chemical Ecology . 16 (3): 861–876. Bibcode :1990JCEco..16..861B. doi :10.1007/BF01016496. PMID 24263601. S2CID 226090.
- ^ "CID 183413". PubChem . Consultado el 12 de mayo de 2012 .
- ^ "Tutorial de SMIRKS". Daylight Chemical Information Systems, Inc. Recuperado el 29 de octubre de 2018 .
- ^ "Reacción SONRISAS y SONRISAS". Daylight Chemical Information Systems, Inc. Recuperado el 29 de octubre de 2018 .
- ^ Lin TS, Coley CW, Mochigase H, Beech HK, Wang W, Wang Z, et al. (septiembre de 2019). "BigSMILES: una notación lineal basada en la estructura para describir macromoléculas". ACS Central Science . 5 (9): 1523–1531. doi :10.1021/acscentsci.9b00476. PMC 6764162 . PMID 31572779.
- ^ Helson HE (1999). "Generación de diagramas de estructura". En Lipkowitz KB, Boyd DB (eds.). Reseñas en química computacional . Vol. 13. Nueva York: Wiley-VCH. págs. 313–398. doi :10.1002/9780470125908.ch6. ISBN . 978-0-470-12590-8.
