Argininemia
| Argininemia | |
|---|---|
| Otros nombres | Deficiencia de arginasa [1] |
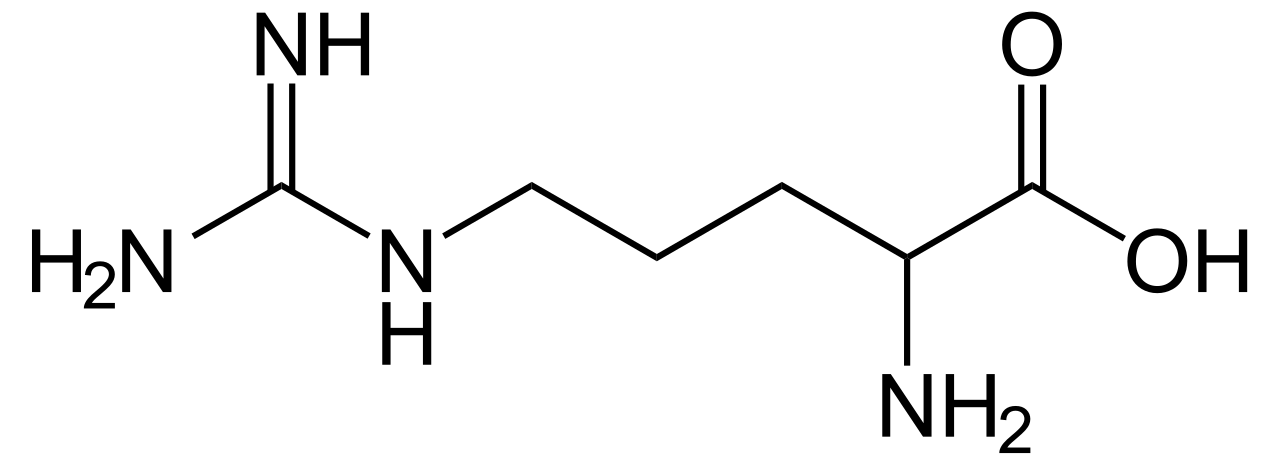 | |
| Arginina | |
| Especialidad | Neurología , genética médica , endocrinología. |
| Síntomas | Letargo, deshidratación [2] [3] |
| Causas | Mutaciones en el gen ARG1 [4] [5] |
| Método de diagnóstico | Concentración de ácido orótico en orina [2] |
| Tratamiento | Ingesta limitada de proteínas, benzoato de sodio [3] |
{kind=link}
La argininemia es un trastorno autosómico recesivo del ciclo de la urea en el que una deficiencia de la enzima arginasa provoca una acumulación de arginina y amoníaco en la sangre . El amoníaco, que se forma cuando las proteínas se descomponen en el cuerpo, es tóxico si los niveles son demasiado altos; el sistema nervioso es especialmente sensible a los efectos del exceso de amoníaco. [2] [6]
Signos y síntomas
La presentación de argininemia, en aquellos afectados, es consistente con lo siguiente: [2] [3]
Genética
{kind=link}

Las mutaciones en el gen ARG1 causan argininemia, que pertenece a una clase de enfermedades genéticas llamadas trastornos del ciclo de la urea . [4] [5] El ciclo de la urea es una secuencia de reacciones que se produce en las células del hígado (hepatocitos). Este ciclo procesa el exceso de nitrógeno , generado cuando el cuerpo utiliza proteínas, y produce urea que se excreta a través de los riñones. [7]
El gen ARG1 proporciona instrucciones para producir una enzima llamada arginasa , que controla los últimos pasos del ciclo de la urea, que produce urea extrayendo nitrógeno de la arginina. [4] En las personas con deficiencia de arginasa, falta arginasa y la arginina no se descompone adecuadamente. En consecuencia, no se puede producir urea y el exceso de nitrógeno se acumula en la sangre en forma de amoníaco . Se cree que el amoníaco y la arginina causan problemas neurológicos y otros síntomas de deficiencia de arginasa. [2]
Esta condición es un trastorno autosómico recesivo, lo que significa que el gen defectuoso está ubicado en un autosoma y se requieren dos copias del gen defectuoso para heredar el trastorno. [6]
Ambos padres de un individuo con un trastorno autosómico recesivo son portadores de una copia del gen, pero generalmente no padecen el trastorno. [6]
Diagnóstico
El diagnóstico de argininemia generalmente se puede realizar mediante una muestra de sangre fetal. [8] Se pueden buscar los siguientes indicadores para detectar la presencia de la afección: [2]
- Concentración de amoniaco en plasma.
- Concentración de ácido orótico en orina
- Actividad de la enzima arginasa en los glóbulos rojos (medición)
Tratamiento
{kind=link}

El tratamiento para las personas con argininemia incluye: [3]
- Benzoato de sodio
- Fenilbutirato de sodio
- Ácido carglúmico
- Fenilbutirato de glicerol
- Palonosetrón
- Clorhidrato de ondansetrón
La pegzilarginasa (Loargys) fue aprobada para uso médico en la Unión Europea en diciembre de 2023. [9]
Referencias
- ^ Herencia mendeliana en línea en el hombre (OMIM): 207800
- ^ abcdef Wong, Derek; Cederbaum, Stephen; Crombez, Eric A. (1 de enero de 1993). "Arginase Deficiency". GeneReviews . PMID 20301338. Consultado el 20 de noviembre de 2016 .actualización 2014
- ^ abcd «Deficiencia de arginasa: antecedentes, fisiopatología, epidemiología». eMedicine . 15 de abril de 2016 . Consultado el 28 de noviembre de 2016 .
- ^ abc "gen ARG1". Genetics Home Reference . Consultado el 28 de noviembre de 2016 .
- ^ ab Ah Mew, Nicholas; Lanpher, Brendan C.; Gropman, Andrea; Chapman, Kimberly A.; Simpson, Kara L.; Summar, Marshall L. (1 de enero de 1993). "Descripción general de los trastornos del ciclo de la urea". GeneReviews . PMID 20301396. Consultado el 20 de noviembre de 2016 .actualización 2015
- ^ abc "deficiencia de arginasa". Genetics Home Reference . Consultado el 20 de noviembre de 2016 .
- ^ Hames, David; Hooper, Nigel (2005). Instant Notes in Biochemistry. Vol. 58 (3.ª ed.). Hoboken: Taylor & Francis Ltd. pág. 408. ISBN 9780203967621. PMID 11098183 . Consultado el 28 de noviembre de 2016 .
{{cite book}}:|work=ignorado ( ayuda ) - ^ Wyllie, Robert; Hyams, Jeffrey S.; Kay, Marsha (2015). Enfermedades gastrointestinales y hepáticas pediátricas. Elsevier Health Sciences. pág. 886. ISBN 9780323370219.
- ^ «Información del producto Loargys». Registro de medicamentos de la Unión . 18 de diciembre de 2023. Consultado el 26 de diciembre de 2023 .
Lectura adicional
- Saudubray, Jean-Marie; van den Berghe, Georges; Walter, Juan H.; Berghe, Georges van den, eds. (2012). Diagnóstico y tratamiento de enfermedades metabólicas congénitas (5ª ed.). Berlín: Springer. ISBN 9783642157202. Recuperado el 28 de noviembre de 2016 .
- Piña-Garza, J. Eric (2013). Fenichel's Clinical pediatric neurology a signs and symptoms approach (7ma ed.). Oxford: Saunders. ISBN 978-1455748129. Recuperado el 28 de noviembre de 2016 .
Enlaces externos
